

Background: Transplant-associated thrombotic microangiopathy (TA-TMA) is an uncommon, but well-recognized, hematopoietic cell transplant (HCT) complication associated with high morbidity and mortality. TA-TMA may result from microvascular endothelial cell (EC) damage induced by HCT conditioning regimens, immunosuppressive drugs, and infections, and it may be mediated by complement activation in some cases. Although there is no consensus on optimal TA-TMA treatment, common interventions include withdrawal, replacement, or dose modification of calcineurin inhibitors (CI) and/or mammalian target of rapamycin (mTOR) inhibitors (ie, CI/mTORi changes). When increased complement activity is present, terminal complement cascade inhibition has been used. Defibrotide (DF) is approved for adult and pediatric patients (pts) with hepatic veno-occlusive disease/sinusoidal obstruction syndrome (VOD/SOS) with renal or pulmonary dysfunction post-HCT in the US and Canada and for pts aged >1 month with severe hepatic VOD/SOS post-HCT in the EU. DF reduces EC activation, promotes fibrinolysis, and may be a novel therapy for TA-TMA. This prospective, open-label, single-arm, multicenter, phase 2 study (EUDRACT: 2018-001101-90) is designed to assess the efficacy and safety of DF for treating pts with high-risk TA-TMA (defined as definite TMA per Postalcioglu M, et al. Biol Blood Marrow Transplant. 2018;24(11):2344-2353) who have failed to respond to CR/mTORi changes.
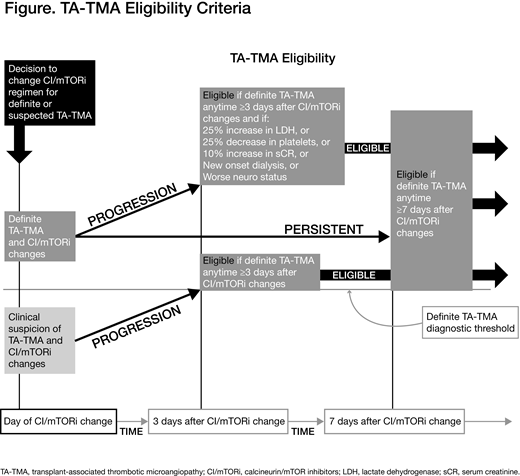
Study Design and Methods: Diagnostic criteria for "definite" TA-TMA include lactate dehydrogenase (LDH) >2 × the upper limit of normal, thrombocytopenia (platelet count <50 × 109/L or ≥50% decrease from peak platelets post-HCT), schistocytes ≥2 per high-power field in peripheral blood, serum creatinine increase >50% from baseline, and a negative Coombs test. Allogeneic HCT pts (aged ≥18 years) who have definite TA-TMA with persistent or progressive disease after 7 or 3 days, respectively, following CI/mTORi changes are eligible (Figure). Pts will be excluded if they relapse after HCT, have grade 4 acute graft-versus-host disease or VOD/SOS, or clinically significant bleeding within 24 hours of enrollment. Planned enrollment is 30 to 40 pts.
Pts will receive a 2-hour intravenous infusion of DF 25 mg/kg/day (6.25 mg/kg every 6 hours) for a recommended duration of 21 days (up to 60 days). Pts will be followed for 180 days from DF start (Day 1).
The study has 2 co-primary efficacy endpoints: hematologic response by Day 45 (relative to DF start) and renal response by Day 45. Hematologic response is defined as normalization of LDH and platelet count ≥50 × 109/L without platelet transfusion for 7 days (if baseline count was <50 × 109/L) or ≥50% increase in platelet count (if baseline count was ≥50 × 109/L). Renal response is defined as <25% increase or decrease in serum creatinine versus baseline at DF start (renal function stabilization) or ≥25% decrease in serum creatinine versus baseline at DF start or independence from dialysis (if pt was dialysis dependent at baseline or started dialysis after baseline) (renal function improvement). Secondary efficacy endpoints are overall survival, duration of hematologic or renal response, proportion of pts achieving hematologic or renal (measured by serum creatinine or estimated glomerular filtration rate) response by Days 60, 90, and 180, independence from dialysis at Days 60, 90, and 180 for pts with dialysis dependence, and neurologic improvement (defined as any increase in Mini-Mental Status Examination score versus baseline or resolution of TA-TMA seizure activity) at Days 7, 14, 21, and 45. Safety endpoints include incidence of treatment-emergent adverse events (AEs), treatment-emergent serious AEs, and AEs of interest (eg, hemorrhage and hypotension) occurring up to 30 days after the last DF dose.
Analysis of the co-primary efficacy endpoints will be evaluated in pts who receive ≥1 DF dose. For each co-primary endpoint, a reference background rate, based upon data from literature and/or registries or research databases, will be established to represent the Day 45 hematologic and renal response rates in a similar pt population in the setting of currently available therapy without DF treatment. A clinically meaningful benefit from DF will be inferred if the lower bounds of the 95% confidence interval of the co-primary endpoints are greater than their respective reference background rates.
Ho:Jazz Pharmaceuticals: Consultancy. Keshelava:Jazz Pharmaceuticals: Employment, Equity Ownership. Swann:Jazz Pharmaceuticals: Employment, Equity Ownership. Tappe:Jazz Pharmaceuticals: Employment, Equity Ownership.
The abstract contains a description of off-label product use. Defibrotide is approved for treatment of hepatic VOD/SOS post-HCT (as described in the background) and this study is evaluating its use in patients with TA-TMA.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal