

Introduction
The majority of patients with Essential Thrombocythaemia (ET) have mutations in JAK2, MPL, and CALR, causing activation of the JAK/STAT pathway; but 10-15% of ET patients lack detectable mutations in these genes, so-called 'Triple Negative' (TN). We applied a systematic approach to investigate mutational status and epigenetic signatures in a cohort of TN ET patients.
Methods and Results
We investigated 46 patients (72% female), median age at diagnosis 35 years (range 8-77 years) including a father and son. All patients were TNusing standard diagnostic assays. We applied deep, error corrected, next generationsequencing (NGS) of 24 genes using the HaloPlexHS platform to peripheral blood samples. Whole exome sequencing was also performed in 23 patients using skin as constitutional control. Overall we identified somatic mutations in 10/46 patients including MPL(3 patients, W515R, W515G, W515S, R537W, VAF 0.02-0.1) JAK2V617F (4 patients, VAF 0.02-0.08); and germline MPLmutations in a further 3 patients (P453R, S505N); including the father and son pair.
We selected patients lacking somatic or germline mutations ("true TN") to analyse gene expression using RNA-seq and DNA methlyation status using 850K Epic Arrays. Patients with JAK2V617Fand CALRmutations and healthy donors (HC) were included as controls.
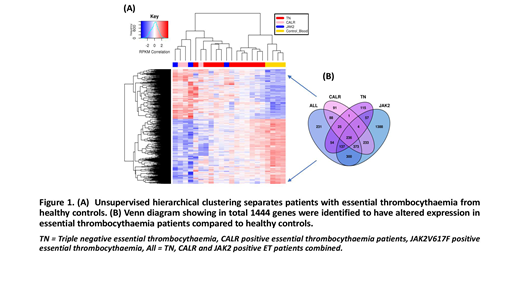
Concerning RNA-seq data, we performed multiple differential analysis of HC vs TN, CALRand JAK2V671F; as well as HC vs all ET samples (adjusted for subtype). Each HC comparison highlighted clear differences between gene expression profiles of HC and disease (Figure 1A). The differentially expressed genes (DEGs) in each comparison overlapped significantly, suggesting that all ET samples have consistent gene expression differences to HC samples regardless of their driver mutation status. In total 1444 differentially expressed genes (ET vs HC) were highlighted (figure 1B). Functional analysis identified significant enrichment for genes involved in the MAPK pathway. Addtionally, we noted upregulation of GATA1,ITGA2B and GP6 genes, not previously reported to be dysregulated in ET.
Correlation of gene expression data with DNA methylation status identified a consistent signature of 306 hypomethylated genes, showing significant enrichment for genes involved in transcriptional misregulation and upregulation of inflammatory regulators such as TNF and NFκB signaling pathways. Next, we identified which transcription factor motifs preferentially bind within these methylation blocks. The blocks showed an enrichment for 6 key transcriptional regulators: ATF3, ATF4, CEBPA, CEBPB, MAX, and RARA. All 6 were significantly upregulated in all ET samples. To validate the motifs, we processed ChIP-seq data from the K562 cell line and identified a significant proportion of the hypo methylated regions are bound by these, transcription factors: 374/410 (91%) regions; 43/410 (10%) are bound by all 6 transcription factors.
Conclusions
A significant proportion (22%) of patients assigned as 'TN' ET via traditional diagnostic techniques in fact harbor known driver mutations at a low allele frequency, suggesting that error corrected NGS approaches may be diagnostically useful in this setting. Additionally, for a group of "true" TN ET patients we demonstrate that patterns of gene expression and DNA methylation are more similar to patients with ET with known driver mutations than healthy controls. Among the upregulated genes are key platelet regulatory genes: GP6, GP1BB, ACTN1and ITGA2B. Furthermore, we identify consistently hypomethylated genes with increased expression across all molecular subtypes of ET which are highly enriched for genes involved in proinflammatory pathways and show that binding of 6 key transcription factorsmay underlie these changes regardless of driver mutation status. Our observations suggest that the ET disease phenotype may, at least in part, be driven by transcriptional misregulation and may be propagated downstream via the MAPK, TNF and NFKappa pathways in addition to activation of JAK/STAT pathways. These findings identify novel mechanisms of disease initiation which require further evaluation.
Dillon:Novartis: Consultancy, Honoraria; Abbvie: Consultancy, Honoraria; Pfizer: Consultancy, Honoraria; TEVA: Consultancy, Honoraria. Mufti:Cellectis: Membership on an entity's Board of Directors or advisory committees, Research Funding; Celgene Corporation: Membership on an entity's Board of Directors or advisory committees, Research Funding, Speakers Bureau. McLornan:Jazz Pharmaceuticals: Honoraria, Speakers Bureau; Novartis: Honoraria. Harrison:Shire: Speakers Bureau; CTI: Speakers Bureau; Celgene: Honoraria, Speakers Bureau; AOP: Honoraria; Janssen: Speakers Bureau; Novartis: Honoraria, Research Funding, Speakers Bureau; Roche: Honoraria; Promedior: Honoraria; Gilead: Speakers Bureau; Sierra Oncology: Honoraria; Incyte: Speakers Bureau.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal