

Background
Regulatory T cells (Tregs) are a non-redundant, suppressive population of CD4+ T cells that are essential for the prevention of autoimmune diseases in humans. They may also contribute to overall immune suppression in malignancies and prevent effective immune surveillance against malignant cells. The correlation between the increased number of Tregs and higher-risk MDS has been shown before (Kordasti, Blood 2007). Expansion of effector T cells (Teff) also signifies the lower-risk disease (Kordasti, BJH, 2009). Using deep phenotyping and unbiased automated clustering we have previously identified novel populations of Tregs with distinct functional properties (Kordasti, Blood 2016), including a CD161-expressing subpopulation (Povoleri, Nature Imm, 2018). The aim of this study was to interrogate Treg and Teff subpopulations in more depth and identify transcriptomic profiles of Tregs and Teff in bone marrow (BM) and peripheral blood (PB) of MDS patients, and potential differences between PB and BM residing cells. Single-cell RNA sequencing (scRNAseq) in combination with oligonucleotide-tagged antibodies and an in-house pipeline utilizing Seurat for QC, data integration and analysis were used for this purpose.
Methods
PB and BM were collected from a low (del(5q)) and a high-risk (monosomy 7) MDS patient. These samples were magnetically enriched for CD4+ T cells, followed by flow sorting of Tregs (CD4+, CD25high, and CD127low) and Teff (CD4+, CD25low, and CD127high). In total, 8 cell fractions were used for downstream scRNAseq and concomitant protein expression profiling using the BD Rhapsody Single-Cell Analysis System (Becton Dickinson, CA, USA).
Cell fractions were first labelled by oligo-tagged antibodies (CD95, CD45RO, CD38, CTLA-4, PD-1, HLA-DR) and a sample tag (for sample multiplexing). After labelling, all samples were combined for cartridge-based single-cell capture and molecular indexing of mRNA transcripts on magnetic barcoded beads. Following bead retrieval and on-bead cDNA synthesis, parallel targeted mRNA (259 T cell-related genes) and Abseq sequencing libraries were generated. Deep sequencing was performed using the HiSeq400 platform.
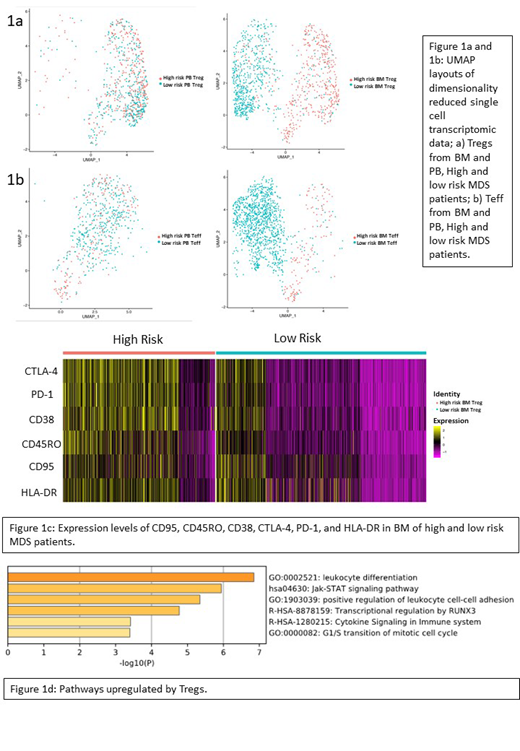
Nonlinear dimensionality reduction was carried out using uniform manifold approximation and projection (UMAP), which produced UMAP layouts and heatmaps to identify populations. Tregs were further stratified according to high/low expression of the CD95 protein, CD161, and CD150 mRNA expression. The Wilcoxon Rank Sum test was used for statistics followed by Bonferroni corrections. The Gene Set Variation Analysis (GSVA) package was used for pathway enrichment analysis.
Results and discussion
Unbiased analysis by UMAP was able to clearly separate the high- and low-risk patients based on the Treg and Teff signature in BM, which was not the case in PB (Figure 1a and 1b).
At protein level, BM Tregs from the high-risk patient had a significantly higher expression of CD95 (p = 7.23 x 10-17), CD45RO (p = 1.06 x 10-51), CTLA4 (p = 4.47 x 10-83) PD-1(p = 5.42 x 10-81), and CD38 (p = 4.16 x 10-63) (Figure 1c), suggesting a more functional Treg profile. GSVA analysis of transcriptomes showed significant changes between high- and low-risk patients, for both Tregs (102 pathways p≤0.05) and Teffs (89 pathways p≤0.05) in BM. Specifically, Tregs from high-risk MDS were more enriched with non-TCR-mediated activation pathways, including leukocyte differentiation, Jak-STAT signaling, and positive regulation of leukocyte cell-cell adhesion (Figure 1d).
A deeper investigation of Tregs showed that subsets of interest, in particular the IL-17 secreting CD161+ Tregs and the hematopoietic stem cell quiescence-maintaining CD150+ Tregs (Hirata, 2018) were found in low abundance in the BM of both high- and low-risk patients; 16% and 10%; and, 1% and 7%, respectively.
In summary, we have developed an analytical pipeline and provide evidence for the ability of simultaneous single-cell transcriptomics and protein expression to identify different BM-specific T cell signatures in high- versus low-risk MDS. Interestingly, Tregs from high risk MDS BM show higher expression of Treg-related proteins and are more enriched in non-TCR-specific activation pathways, which may reflect a myeloid derived inflammatory environment. Overall, these findings suggest a bone marrow-specific Treg signature in MDS, which could distinguish low- and high-risk disease.
McLornan:Jazz Pharmaceuticals: Honoraria, Speakers Bureau; Novartis: Honoraria. Platzbecker:Celgene: Consultancy, Honoraria; Novartis: Consultancy, Honoraria; Abbvie: Consultancy, Honoraria. Kordasti:Celgene: Research Funding; Novartis: Research Funding; Boston Biomed: Consultancy; API: Consultancy.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal