

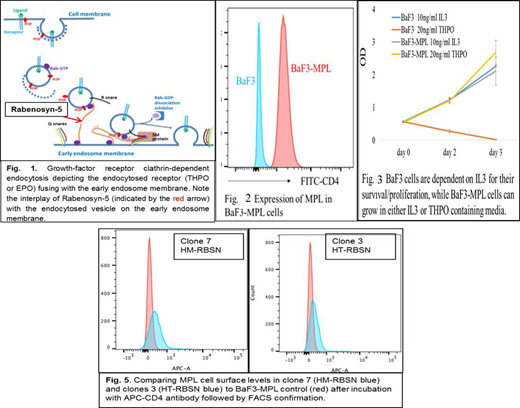
Background: Myeloproliferative neoplasms (MPNs), commonly polycythemia vera (PV), primary myelofibrosis (PMF) and essential thrombocythemia (ET), are a group of malignant hematologic disorders characterized by proliferation and accumulation of terminally differentiated blood cells of erythroid, myeloid and megakaryocytic origin (Cazzola, Blood 2014).Somatic driver mutations, frequently JAK2V617F, MPLW515L/K or mutant CALR in sporadic MPNs affect signaling by the thrombopoietin (THPO) and/or the erythropoietin (EPO) receptor (Pickman, PLoS 2016). MPL is the receptor of THPO and JAK2 is the key downstream mediator of THPO/MPL signaling (Nangalia NEJM 2013). In familial MPNs (families with more than 1 member developing a MPN) a germline mutation predisposes to disease development. These mutations can reveal pathways or processes involved in the pathogenesis of the disease, independent of, or cooperating with, the somatic driver mutations (Rumi, JCO 2007). We identified 3 putative predisposing germline mutations in 1 family, 3 affected generations developed MPN (PMF, ET and PV): RBSN frameshift,JAK2R340Q & PRKAA2Q425P missense variants. RBSN encodes Rabenosyn-5 protein (RBSN) which plays a role in early growth-factor receptor clathrin-dependent endocytosis (Nielsen, J Cell Biol 2000) (Fig.1).We discovered that wild type (WT) Rabenosyn-5 regulates cell surface MPL levels in hematopoietic progenitor cells and predict that MPL levels on the cell surface are negatively associated with WT Rabenosyn-5 levels. We hypothesize that the RBSN mutation predisposes to MPN development in this family, leading to abnormal activation of THPO signaling by reducing MPL turnover.
Methods: Peripheral blood DNA from patient samples were analyzed for potential putative mutations using Agilent SureSelect Human ALL Exon V5+UTRs exome capture kit followed by parallel sequencing with Illumina HiSeq 2000. In vitro studies involved using the BaF3 system and CRISPR-Cas9 gene editing technique. We generated BaF3-MPL (BaF3 cells transduced with human CD4-MPL fusion) cell lines that rely on IL3 or human THPO cytokines for growth & proliferation (Fig.2&3). We transduced RBSN into the BaF3-MPL cell lines and produced RBSN heterozygous (HT) and homozygous (HM) mutants. We first generated LentiCRISPRV2GFP-mRBSN vector by inserting RBSN specific guide RNA into the LentiCRISPRV2GFP vector and generated lentivirus expressing both Cas9 and guide RNA by co-transfecting 293T cells with LentiCRISPRV2GFP vector and viral packaging vectors. The final virus was used to infect BaF3-MPL cells. Transduced cells were purified by FACS to select GFP+ cells and individual cells were grown in culture. RBSN gene mutations were examined using PCR and sequenced accordingly. To study whether the RBSN mutation alters the surface MPL levels, we stained BaF3-MPL, HT-RBSN-BaF3 and HM-RBSN-BaF3 cells with APC-CD4 antibody and the surface levels of MPL were detected by FACS by comparing the mean fluorescence intensity (MFI) of APC.
Results: We identified several colonies with either HT-RBSN-BaF3 or HM-RBSN-BaF3. The guide RNA we designed specifically targeted the 12th exon of the RBSN gene which best recapitulates the mutations observed in our patients, thus, the frameshift mutations in HT-RBSN-BaF3 and HM-RBSN-BaF3 cell lines we predict will make truncated forms of RBSN which will be verified by Western Blotting for future experiments. Surface levels of MPL in the HT-RBSN-BaF3 and HM-RBSN-BaF3 cells are greater than that in BaF3-MPL cells, suggesting RBSN negatively regulates THPO-MPL signaling by regulating the cell surface MPL levels (Fig. 4).
Conclusions: RBSN is a regulator of receptor trafficking, which mediates the fusion of the endocytosed receptor to the early endosome and then lysosome for degradation. We propose that genetic inactivation of RBSN might prevent the endocytosed MPL from endosome-degradation and enhance the reuse of MPL by recycling. Future experiments are planned to include in vivo studies to study whether inactivation of RBSN promoted MPN-like disease development into NSG mice and transducing our established cell lines to label individual cell compartments to study THPO-stimulated signaling. Our findings, if confirmed, will further clarify aspects of familial and somatic MPN pathogenesis and may inform efforts to devise new therapeutic and diagnostic strategies for this disease complex.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal