

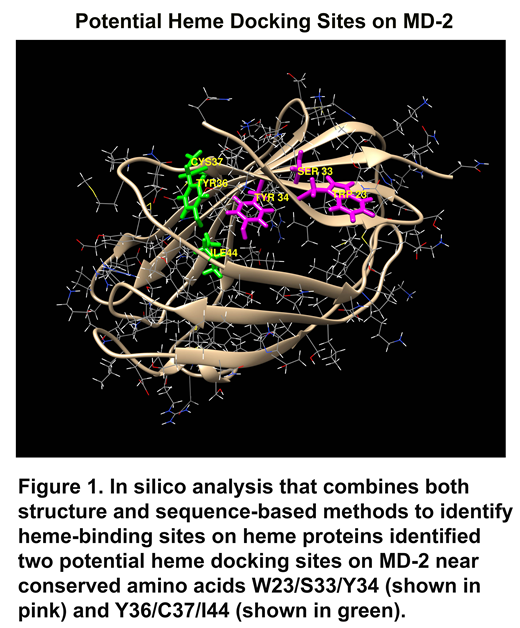
Lipopolysaccharide (LPS), the first-identified TLR4 agonist, binds myeloid differentiation factor-2 (MD-2) in association with TLR4 to initiate TLR4 signaling. LPS binds to a large hydrophobic pocket in MD-2 and directly bridges the MD-2/TLR4 heterodimer. The MD-2/TLR4 complex also recognizes a diverse number of endogenous molecules released from injured cells called damage-associated molecular patterns or DAMPs. One such DAMP is heme. Large amounts of heme can be released intravascularly by trauma, sepsis, malaria and red blood cell disorders such as sickle cell disease (SCD). Recent studies underscore the importance of heme-mediated MD-2/TLR4 activation in inflammation, vessel occlusion, lethality and pulmonary injury in SCD. Therefore, we examined human MD-2 for potential heme activation sites. Recombinant MD-2 (rMD-2) was produced by transfecting Chinese hamster ovary (CHO) cells with human MD-2 plasmids. After 72 hours, Western blots of the CHO-conditioned media demonstrated soluble rMD-2 was present. Heme was shown to bind rMD-2 using pull-down assays utilizing heme-agarose or biotin-heme with streptavidin-agarose coupled with MD-2 Western blots of the pellet. These pull-down assays of rMD2 were inhibited by excess heme, indicating specific binding of heme to rMD-2. UV/visible scanning spectroscopy (250 - 550 nm) of purified rMD-2 in the presence or absence of heme, confirmed specific rMD-2-heme binding. In silico analyses combining both structure and sequence-based methods, identified two potential heme docking sites on MD-2 near conserved amino acids W23/S33/Y34 and Y36/C37/I44 (Figure 1). To determine whether MD-2 mutations at these two sites affect heme-MD-2/TLR4 signaling, HEK293 cells were transfected with plasmids encoding human MD-2, TLR4, CD14 and an NF-κB luciferase reporter. After 24 hours, transfected cells were stimulated with heme (10 μM) or LPS (10 ng/ml) for 6 hours and NF-κB luciferase reporter activity was measured. Heme or LPS treatment elicited robust luciferase activity. The addition of both heme and LPS had an additive effect on NF-κB luciferase activity. Absence of an MD-2, TLR4 or CD14 plasmid abolished NF-κB luciferase reporter responses to heme and/or LPS. When plasmids encoding MD-2 point mutants W23A or Y34A were introduced into MD-2, heme-induced NF-κB luciferase activity was inhibited 91-92% compared to WT-MD-2. The S33A MD-2 mutant stimulated NF-κB luciferase activity by 40%. NF-κB activation by LPS was marginally affected by the same mutants. Biotin-heme/streptavidin-agarose pulled down 68% less W23A mutant MD-2 and 80% less W23A/S33A/Y34A mutant MD-2 than WT-MD-2. In contrast, at the other potential heme binding site, heme-induced NF-κB luciferase activity was increased in mutants Y36A (120%), C37A (121%) and I44A (230%) compared to WT-MD-2. These data suggest that amino acids W23 and Y34 on MD-2 are specific for heme binding and TLR4 signaling. This heme activation site was targeted for potential inhibitors using virtual screening. The virtual screen identified 60 potential inhibitors for screening in heme-stimulated primary human umbilical vein endothelial cells (HUVEC) and a human U-937 monocyte cell line. Four of these molecules inhibited Weibel-Palade body P-selectin and von Willebrand factor expression in HUVEC and IL-8 secretion by U-937 cells stimulated with heme. We conclude that heme activates MD-2/TLR4 signaling at residues W23 and Y34 on MD-2, which might be a drugable target in SCD and other hemolytic diseases.
Belcher:Mitobridge, an Astellas Company: Consultancy, Research Funding. Vercellotti:Mitobridge, an Astellas Company: Consultancy, Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal