

Introduction: Despite remarkable clinical efficacy, CAR-T therapy has been limited by life threatening toxicities in over 30% of patients.1, 2 Toxicities primarily manifest as cytokine release syndrome (CRS) characterized by an early phase with fever, hypotension and elevations of cytokines including IFNγ, GM-CSF, TNF, IL-10, and IL-6 and a later phase associated with life-threatening or life-ending neurologic events. We hypothesized that reversible inhibition of CAR mediated signaling will enable controllable regulation of CAR-T cell activity in vivo and mitigate CAR-T mediated toxicities. There are specific protein kinases such as the SRC kinases, LCK, and ZAP-70 that are known to be involved in various cellular signaling pathways, especially T cell receptor mediated signaling and may also be appropriate targets for modulating (both enhancing and inhibiting) CAR-T function in a rapid and reversible fashion in vivo. Our hypothesis is that small molecule inhibitors of TCR signaling and downstream pathways could be identified using specific high throughput screens.
Methods: To identify novel inhibitors of CAR-T cell proliferation, we developed a high throughput kinase inhibitor screen to identify compounds that reversibly inhibit CAR-T function. T cells containing a third generation CAR targeting CD19 cells (CAR19) and CD19+ tumor cells (Ramos cells expressing both GFP and luciferase) were incubated at an effector to target ratio of 1:1 in 96 well plates in the presence of 1µM of each inhibitor. After 24 hours, tumor cell death induced by CAR-T was measured using bioluminescence (BLI) imaging. Small molecules that inhibited CAR-T proliferation and cytotoxicity were determined by assessing the BLI signal in each well.
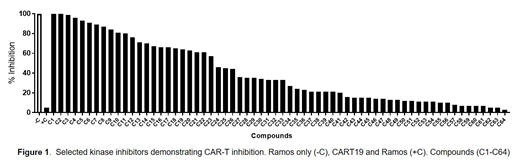
Results: A protein kinase inhibitor library (Selleckchem, Texas) containing 644 independent compounds was tested (Figure 1). Of the 644 kinase inhibitors tested, 32 were found to be potent inhibitors of CART19 cell activation and cytotoxic killing of CD19+ target tumor cells, reducing anti-tumor viability in 24 hours by >50% compared to vehicle control. Compounds such as Nintedanib (C3), Dasatinib (C9), and Saracatinib (C12), all SRC kinase inhibitors, were able to inhibit cell killing by 99%, 84%, and 76% respectively. Next we assessed the reversibility of CAR-T cell mediated killing upon removal of inhibitors from the cultures. Reinitiation of potent, anti-tumor activity was observed within 24 hours after inhibitor removal, confirming reversible nature of CAR-T cell inhibition by the three most potent compounds.
Conclusions: Recent publications (Weber et al Blood Adv, 2018, Westermann et al. Sci Transl Med, 2019) have also shown that dasatinib can reversibly suppresses CAR-T cell cytotoxicity, cytokine secretion, and proliferation in vitro and in vivo. 3, 4 Here we confirm the reports of others regarding dasatinib and that show for the first time that reversible inhibition of CAR-T activity by kinase inhibitors is not limited solely to dasatanib, but is observed with other small molecules targeting many different kinases. This work further demonstrates the potential applications of tyrosine kinase inhibitors as a safety switch to modulate CAR-T cell toxicity.
1. Maude, NEJM 2014
2. Davila, SciTransMed 2014
3. Mestermann SciTransMed 2019
4. Weber. Blood Adv 2019
Cooper:Wugen: Consultancy, Equity Ownership, Patents & Royalties. DiPersio:WUGEN: Equity Ownership, Patents & Royalties, Research Funding; Magenta Therapeutics: Equity Ownership; Celgene: Consultancy; Karyopharm Therapeutics: Consultancy; RiverVest Venture Partners Arch Oncology: Consultancy, Membership on an entity's Board of Directors or advisory committees; Cellworks Group, Inc.: Membership on an entity's Board of Directors or advisory committees; NeoImmune Tech: Research Funding; Bioline Rx: Research Funding, Speakers Bureau; Macrogenics: Research Funding, Speakers Bureau; Incyte: Consultancy, Research Funding; Amphivena Therapeutics: Consultancy, Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal