

The BCL2 Gly101Val mutation may be acquired in patients with chronic lymphocytic leukaemia (CLL) treated with venetoclax (VEN), leading to reduced drug binding affinity and secondary resistance. In the majority of patients, the Gly101Val mutation is subclonal within the CLL compartment consistent with the presence of alternative resistance mechanisms in CLL cells not harboring the Gly101Val mutation. To date, two Gly101Val mutated patients have been identified with co-existing candidate resistance mechanisms in Gly101Val non-mutated cells; one with BCL-XL over-expression (Blombery et al, Cancer Discov., 2019) and another with a second subclonal candidate BCL2 resistance mutation - Asp103Tyr (Tausch et al, Haematologica 2019). Given the possibility of additional resistance mechanisms, we investigated patients with progressive CLL on VEN harboring the Gly101Val mutation for the presence of additional acquired resistance mutations in BCL2.
Ten patients with progressive CLL on VEN with Gly101Val mutations were identified by sensitive allele-specific droplet digital PCR. To further assess for alternative BCL2 mutations in this cohort we performed ultra-deep amplicon-based next generation sequencing (NGS) (median depth ~50,000X) targeting BCL2. An amplicon variant caller (Canary) specifically designed for low level variant calling was used (Doig et al, BMC Bioinformatics, 2017). To achieve enhanced specificity we performed digital NGS with PCR error-correction using unique molecular indexes (UMI) (QiaSEQ Targeted DNA Panel). Given the high GC content of BCL2 we also used hybridization-based NGS using a custom targeted panel (Blombery et al, BJH 2017) combined with a sensitive unpaired variant caller (GATK4/Mutect2).
In 7/10 (70%) patients, BCL2 mutations in addition to the Gly101Val were detected. Recurrent mutations (detected in more than one patient) were Asp103Tyr, Asp103Glu, Arg107_Arg110dup, and Val156Asp. All additional recurrent mutations were confirmed to be absent prior to commencing VEN (sensitivity 1% variant allele frequency[VAF]). Phase-analysis of NGS reads was consistent with the presence of the additional recurrent mutations on different alleles (and therefore cells, assuming heterozygosity) to both each other and to Gly101Val. Multiple addition recurrent mutations were observed in patients in the cohort with one patient harboring three recurrent mutations in addition to the Gly101Val (Asp103Tyr, Asp103Glu, Val156Asp). In multiple patients in the cohort, the VAF of non-Gly101Val mutations exceeded that of the Gly101Val mutation. Importantly, in all patients a significant (albeit variable) proportion of CLL cells were found to be BCL2 wild-type consistent with the presence of as yet unidentified resistance mechanisms unrelated to BCL2 mutations. In one patient, two additional non-recurrent mutations were observed (Ala113Gly and Arg129Leu) in addition to Gly101Val and Val156Asp. Again, all four mutations in this patient were observed to be in mutually exclusive NGS reads.
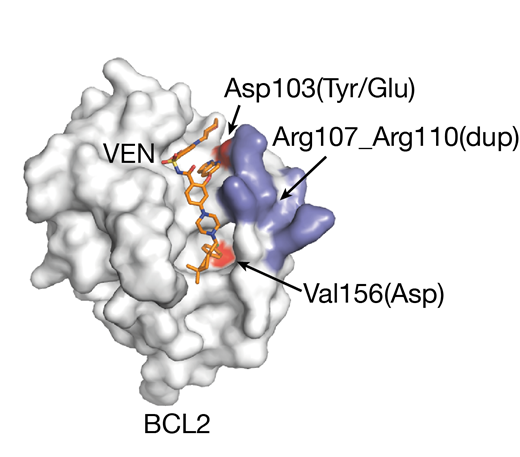
Strikingly, all of the recurrent acquired BCL2 mutated residues identified in our cohort are situated in the BCL2 binding groove that binds VEN (Figure 1). The Asp103 codon in the P4 pocket is critical for VEN binding through hydrogen bonding between its sidechain and the azaindole moiety of VEN. The Asp103Glu mutation is noteworthy given that the equivalent residue to Asp103 in BCL-XL is a Glu, which reduces VEN binding to BCL-XL. The Val156 mutation situated at the base of the P2 pocket is close to the chlorophenyl moiety of VEN and a change to Asp in this position may disrupt VEN binding. Ongoing binding experiments and modeling in cellular systems will further elucidate the mechanism and contributions of these new recurrent mutations to VEN resistance.
In summary, we have extended the landscape of acquired candidate resistance mutations occurring in patients treated with VEN to include four novel recurrent BCL2 mutations. Moreover, our data are consistent with the emerging observation of multiple acquired resistance mechanisms operating in different CLL cells in a single patient contributing to an "oligoclonal" pattern of clinical relapse on VEN therapy.
Figure 1 - BCL2 protein structure surface bound to venetoclax (VEN) in orange. The Asp103Tyr, Asp103Glu and Val156Asp mutation sites are shown in red and Arg107_Arg110dup region in blue
Blombery:Janssen: Honoraria; Invivoscribe: Honoraria; Novartis: Consultancy. Anderson:Walter and Eliza Hall Institute: Employment, Patents & Royalties: Institute receives royalties for venetoclax, and I receive a fraction of these.. Seymour:Acerta: Consultancy; Celgene: Consultancy, Research Funding, Speakers Bureau; Janssen: Consultancy, Research Funding; AbbVie: Consultancy, Honoraria, Research Funding, Speakers Bureau; Roche: Consultancy, Research Funding, Speakers Bureau; Takeda: Consultancy. Huang:Genentech: Patents & Royalties: DCSH is an employee of the Walter and Eliza Hall Institute which receives milestone and royalty payments related to venetoclax. Roberts:AbbVie: Other: Unremunerated speaker for AbbVie, Research Funding; Australasian Leukaemia and Lymphoma Group: Membership on an entity's Board of Directors or advisory committees; Janssen: Research Funding; Walter and Eliza Hall Institute: Patents & Royalties: Institute receives royalties for venetoclax, and I receive a fraction of these.; BeiGene: Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal