

Introduction: Juvenile myelomonocytic leukemia is a heterogenous disease of early childhood that has both myelodysplastic and myeloproliferative properties. Outcomes range from spontaneous resolution with little to no therapy in some patients while others relapse even after hematopoietic stem cell transplantation. However, there is no single biomarker reliably capable of predicting outcome.
Patients and Methods: Our three groups have recently published data regarding the predictive capacity of DNA methylation profiling using the Illumina Infinium Methylation 450k platform. Methylation data along with clinical annotation from 256 JMML patients (126 EWOG/MDS, 93 Japanese and 37 USA) were analyzed and a consensus algorithm to determine DNA methylation clustering was agreed upon. A technical validation of two clinically oriented DNA methylation approaches was undertaken using DNA from 32 patients (12 EWOG/MDS, 5 Japan and 15 USA) included in the previous publications. A biological validation using DNA from additional 48 JMML patients (9 EWOG/MDS, 19 Japan and 20 USA) that were not previously tested was also undertaken to independently validate the DNA methylation subgroups. All 80 patients from the technical and biological cohorts were analyzed for genetic mutations using a custom 25-gene panel next-generation-sequencing (NGS) approach. Informed consent to use of patient material was obtained in accordance with the Declaration of Helsinki and approved by the local institutional review boards.
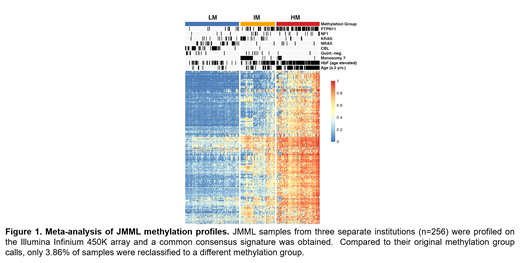
Results: Unsupervised hierarchical consensus clustering of 256 JMML patients using the 5000 CpG sites with the highest standard deviation separated patients into three distinct methylation subgroups using the 450k data. Based on the mean beta-value detected, the subgroups were designated as low methylation (LM), intermediate methylation (IM) and high methylation (HM). Strikingly, when comparing our new DNA methylation subgroup assignments to the published 450k DNA methylation subgroups, only 11 patients (3.86%) were now re-classified into a different methylation subgroup (Figure 1). Next two approaches were taken with the intent of clinical grade implementation of DNA methylation testing. In one approach at EWOG/MDS, a multiclass classification model (XGBoost) was developed based on the 5000 CpGs and tested by classifying the validation cohort into the respective methylation subgroups. When analyzing the relative contribution of each CpG to the model performance (Model Gain), we identified 10 CpG sites which contributed most to the model. Illumina Infinium 850k array testing using XGBoost of the technical cohort provided an accuracy of 90.6% resulting in misclassification of 3/32 patients as compared to the original 450k based designation of methylation subgroups. In the USA, an amplicon based, bisulfite treated, NGS approach (Methyl-NGS) using primers to sequence the top 3000 CpG sites from the meta-analysis was tested in the technical and biological validation cohorts. Using a standard deviation of 0.25 of the most variable probes to determine the methylation clusters, there was 100% concordance between the 450k and the Methyl-NGS designation of methylation subgroups.
Conclusions: In this single largest collection of JMML data ever presented, we demonstrate the reproducibility of DNA methylation as a predictive biomarker of outcome across continents, platforms and different patient cohorts. The DNA methylation consensus subgroups generated from 256 JMML patients were associated with previously recognized variables such as age, platelet count, fetal hemoglobin status and the number of genetic alterations at diagnosis. However, DNA methylation cluster designation provided additional prognostic information, in particular in young patients or patients with monosomy 7 where fetal hemoglobin is not interpretable and clarified that not all patients with PTPN11, KRAS or NF1 are classified as IM or HM as might be expected based on genetics alone. Clinical grade implementation of DNA methylation testing using either Infinium based XGBoost or Methyl-NGS both provided a high degree of reproducibility with the original 450k data. Combining DNA methylation with previously recognized variables allows for a robust risk-stratification algorithm that is now ready to be implemented in clinics around the world and integrated into clinical trials.
Loh:Medisix Therapeutics, Inc.: Membership on an entity's Board of Directors or advisory committees. Niemeyer:Celgene: Consultancy. Lipka:InfectoPharm GmbH: Employment.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal