

Primary myelofibrosis (PMF) is the most aggressive Philadelphia-negative myeloproliferative neoplasm. Heterogeneity in disease phenotype and outcome led to investigate the influence of germline host genetic variations on disease onset and evolution. We recently found that patients with secondary myelofibrosis are enriched in the polymorphic allele variant of the -2518 A/G SNP of the Monocyte Chemoattractant Protein-1 (MCP-1), and that this SNP correlates with disease severity (Masselli E. et al, Leukemia 2018). Here, we examined the interactive effects of MCP-1 allele variants with the baseline phenotype in PMF, and how these variants impact the disease progression.
MCP-1 -2518 SNP genotyping was performed by TaqMan Predesigned SNP Genotyping Assays on DNA extracted from peripheral blood granulocytes of 201 PMF patients and 249 matched local control subjects (CTRL).
One-hundred and nineteen out of 201 PMF were males (59.2%). Median age was 52y (18-80y). N.103 patients (51.2%) were pre-fibrotic PMF. Driver mutations occurred as follows: 123 (61.2%) harbored a JAK2V617F mutation, 47 (23.4%) had CALR mutations, 20 (9.9%) were triple negative and n.8 (4%) had MPL mutations. Three patients had both JAK2V617F and CALR mutation. After a median follow-up of 84.7 months, 37 patients (18.4%) died, 38 (18%) experienced blast transformation (BT) and 23 patients (11.4%) received allogeneic hematopoietic stem cell transplantation (ASCT). Median age of CTRL was 64y (28-85y), and 132/249 (53%) were males.
Consistently with our previous report, PMF displayed similar genotypic and allelic frequencies of the local control population (O.R., 1.04). The G allele distribution was also similar in JAK2V617Fpos vs. JAK2V617Fneg PMF and did not correlate with the allele burden (O.R., 1.29 and 1.51, respectively).
After verifying that PMF with only 1 copy of the risk allele (A/G), had a similar risk of the reference group with 0 copies (A/A), we used a recessive genetic model to compare PMF with 2 copies of the risk variant (G/G, hereby referred as the MCP-1 high-risk group) vs. A/A+A/G PMF (the MCP-1 low-risk group). Genotype-phenotype correlations with disease parameters at the time of diagnosis were performed by χ2/Fisher exact test.
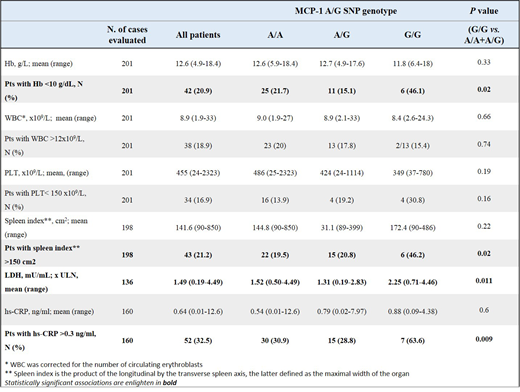
We found that the MCP-1 high risk group displayed a significantly higher frequency, at the time of diagnosis, of severe anemia, massive splenomegaly, elevated LDH and higher levels of hs-CRP (high-sensitivity C-reactive protein) (see Table). These results indicate that MCP-1 high-risk variant has a role in determining the disease phenotype and the pro-inflammatory microenvironment of PMF.
Additionally, we assessed whether the MCP-1 high-risk variant could predispose the acquisition of risk factors in PMF. On the basis of the recessive genetic model, we found that the MCP-1 high-risk group was about four times as likely to progress to leukocytosis as was the reference group (HR, 4.16; 95% CI; 1.37 to 12.5; P=0.011).
To weight the clinical relevance of our findings, we tested whether the MCP-1 high-risk variant might affect the disease progression. ASCT occurrence resulted significantly higher in the high-risk group as compared to the low-risk (HR = 3.22; 95% CI, 1.06 to 10; P=0.038). In the composite endpoint model that considered death for any cause, BT or ASCT, PMF carrying the high-risk genotype presented a shorter time to event as compared to the low-risk group (HR, 2.56; 95% CI, 1.19 to 5.55; P=0.016).
In conclusion, the MCP-1 SNP appears to identify PMF patients at higher chance to present with adverse hematological features and more likely to progress rapidly to either death, BT or ASCT.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal