

Introduction: FASN catalyzes de novo fatty acid (FA) biosynthesis, which is an oncogenic function observed in many cancers, including NHL. HIF1a inducible FASN activity is responsible for overcoming negative hypoxic influence and upregulation of glucose metabolism as observed during premalignant transformation. FASN catalytic activity is also dependent on precursors derived from glucose metabolism. However, implications of FASN targeted therapies on these interdependent metabolic interactions and the overall impact on cancer cell proliferation remains unknown.
Methods: FASN small molecule inhibitors cerulenin, orlistat, TVB3657 and TVB3166 were evaluated using a diverse panel of B cell NHL lines and primary NHL cells for the impact on FASN inhibition signaling & induction of cell death (MTT, caspases & AnnexinV/PI). Global transcriptomics were done with Affymetrix Human 2.0 ST Genechip with Gene Set Enrichment and Ingenuity Pathway Analysis (GSEA & IPA). Metabolomic profiling was performed using mass spectrometry. TXNRD1, GSR, NQO1 expression and activity were evaluated by western blot and using enzyme assay kits. Global DNA & RNA synthesis were evaluated using ClickIt Edu and EU assay kits. RNAseq data available from 775 NHL patients were utilized for metabolic transcriptome mapping.
Results: Treatment with all FASN inhibitors resulted in dose- and time-dependent reduction (>90%) in cell viability and cell death in all NHL cell lines. GSEA and IPA identified cell cycle & RNA metabolism as downregulated biological processes with carbohydrate and oxidative stress as upregulated biological processes observed. "Key gene" analysis predicted TNF signaling as a prominent response to FASN inhibition, validated as increased TNF secretion by ELISA with cell fractionation and western blot analysis revealing activation of TNF-PI3K signaling. Activation of PI3K with FASN inhibition also corresponded with increased expression of PI3K-dependent metabolic genes associated with glucose and lipid metabolism. Furthermore, metabolomic profiling in SUDHL10 cells revealed accumulation of the FASN precursor acetyl-CoA with FASN inhibition that was accompanied by increased ketogenic glycolytic and citric acid cycle activity.
In addition, NADPH accumulation (FASN substrate) occurred with FASN inhibition, which was accompanied with reduction in ribose-phosphate and nucleotide pools as these processes are relevant to NADPH-generating PPP function. G6PD, TXNRD1, GSR & NQO1 were identified as "key genes" responsive to FASN inhibition. Interestingly, this cluster of "key genes" represented the entire enzymatic activity related to the first rate-limiting step in PPP. Subsequently, we determined that increased NADP(H) pools were responsible for impaired NADPH regenerating functions of TXNRD1 and GSR antioxidant enzymes, with PI3K-dependent activation of NQO1 and uptake of glucose facilitating de novo glutathione synthesis, which substituted for the loss of antioxidant functions in SUDHL10, SUDHL4 and Raji cells. All responses were sensitive to inhibition by PI3K inhibition (e.g., BKM120) with co-targeting via FASN & PI3K inhibition resulting in markedly increased oxidative stress, loss of mitochondrial membrane potential & synergistic cell death in NHL cell lines and primary NHL cells.
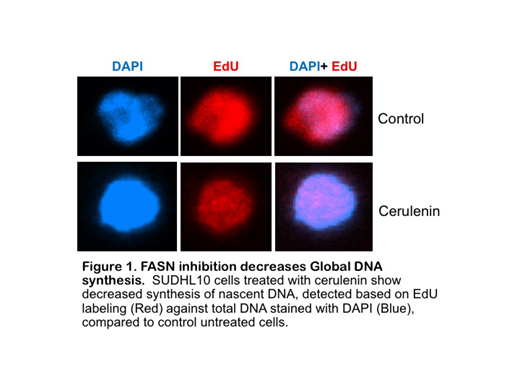
Finally, FASN inhibition was associated with reduction in ribo/deoxy-ribonucleotide pools that decreased global transcriptional activity (de novo RNA synthesis, by EU labeling) and replication (by EdU incorporation in DNA) (Fig 1). Analysis of RNAseq data and mapping of metabolic transcriptome from 772 NHL patients showed consistently elevated expression of genes related to glycolysis, citric acid cycle, fatty acid and nucleotide metabolism in patients with mutations in p53, MYC, BCL2, mTOR, MYD88, PIM2 & CREBP genes, suggesting that these onco-metabolic interactions may be important for lymphomagenesis.
Conclusions: Taken together, FASN oncogenic activity appears to extend beyond de novo fatty acid biosynthesis, serving as a central onco-metabolic regulator of malignant cell proliferation vis-à-vis integrating glucose, nucleotides, and antioxidant metabolic functions in NHL. In addition, co-targeting FASN and PI3K induced synergistic cell death. Altogether, blocking FASN and the dependent onco-metabolic functions represent highly novel targets for therapeutic strategies in NHL.
Chen:Oncomics, LLC: Consultancy, Patents & Royalties. Dave:Data Driven Bioscience: Equity Ownership. Evens:Seattle Genetics: Consultancy, Honoraria, Research Funding; Pharmacyclics: Consultancy, Honoraria; Epizyme: Consultancy, Honoraria; Tesaro: Research Funding; Verastem: Consultancy, Honoraria.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal