

Background
As a single platform, RNA sequencing (RNA-Seq) can accurately identify majority of oncogenic driver fusion genes and aneuploidy. This provides refined molecular subtyping and risk stratification; RNA-Seq is increasing used upfront for genetic subtype assignment in on-going contemporary ALL trials. The IGH gene is highly expressed in our diagnostic ALL samples using RNA-Seq. We investigated the usefulness of RNA-Seq in identifying IGH disease sequences for MRD monitoring in 258 childhood B-lymphoblastic leukemia (B-LL) samples.
Methods
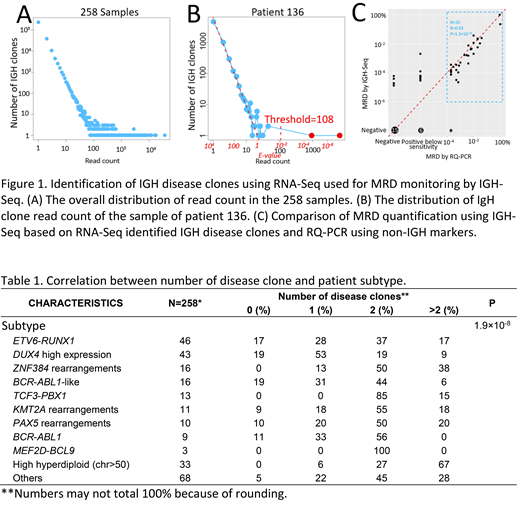
RNA-seq was performed on a total of 259 diagnostic bone marrow or peripheral blood samples from children with B-LL treated in the Ma-Spore ALL 2003 and ALL 2010 studies. The reads were aligned to germline IGH gene V, D and J segments, and reads covered IGH VHDJH or DJH junctional regions are clustered into clones. Overall, the distribution of read count of the IGH background clones follows Zipf's law, i.e. the read count of the IGH background clones is negative linearly correlated with the number of IGH clones with that read count in the log-log scale (Figure 1A). IGH disease clones are outliers with exceedingly high read count. For each sample, a linear model is fitted between log transformed read count (using only read count ≤10) and log transformed number of IGH clones. The expected number of IGH clones (E-value) of a read count can be predicted by the fitted linear model. A clone with E-value <0.01 is defined as a disease clone (Figure 1B). Disease clones identified by RNA-Seq are compared with conventional Sanger sequencing.
Results
In 90.3% of the patients, 497 IGH disease clones (median 2, range 0-7 clones/patient) are identified. The number of IGH disease clones significantly correlated with genetic subtype (P=1.9×10-8; Table 1). Specifically, high hyperdiploid patients have the most IGH disease clones (median 3) with 67% (22/33) have >2 IGH disease clones, due to the gain of chromosome 14. In contrast, DUX4 high expressers have the least (median 1) IGH disease clones with 19% (8/43) have no IGH disease clone, due to the rearrangements involving one of the IGH locus.
Of the 348 IGH disease clones identified by Sanger sequencing, 90.8% are also identified by RNA-Seq; and in the 199 Sanger sequences that yielded sensitive markers, 191 (96.0%) are also defined as IGH disease clones by RNA-Seq. In addition, RNA-Seq identified 43% more IGH disease clones. In 69 patients lacking sensitive IGH targets by Sanger sequencing, the disease clones identified by RNA-Seq were used collectively for MRD monitoring by targeted deep sequencing of IGH (IGH-Seq) and showed high correlation with conventional RQ-PCR MRD using non-IGH targets (R=0.93; P=1.3x10-14;Figure 1C). In addition, IGH-Seq showed improved sensitivity with quantifiable MRD in 20% more samples compared to RQ-PCR (78% vs 58%; Figure 1C). In the samples that are positive below sensitivity or negative by RQ-PCR, IGH-Seq obtained quantifiable MRD for 60% (9/15) or 25% (5/20) of the samples (Figure 1C).
Conclusion
In conclusion, IGH disease clone sequences are highly expressed and can be distinctly identified from whole transcriptome RNA-Seq in >90% of children with B-LL. These can be used for molecular MRD monitoring using RQ-PCR or NGS-based IGH-Seq. In addition to identifying the constellation of oncogene fusions and chromosomal aneuploidies that drive B-LL and RNA-Seq can identify IGH MRD sequences for molecular MRD monitoring.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal