

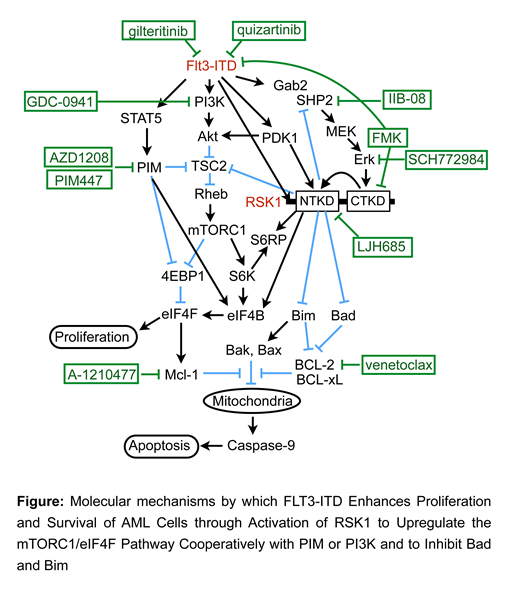
FLT3-ITD is the most frequent tyrosine kinase mutation in acute myeloid leukemia (AML) associated with poor prognosis. We previously reported that FLT3-ITD activates the mTORC1/S6K/4EBP1 pathway cooperatively through the STAT5/PIM and PI3K/Akt pathways in AML cells, thus promoting proliferation and survival as well as therapy resistance at least partly by enhancing the eIF4F complex-mediated cap-dependent translation of growth- and survival-related mRNAs. The RSK family serine/threonine protein kinases, including the predominant isoforms expressed in AML RSK1 and RSK2, posses the N- and C-terminal kinase domains (NTKD and CTKD); CTKD is activated by Erk and recruits PDK1 to activate NTKD, which phosphorylates downstream substrates to promote cell growth and survival through regulation of translation as well as transcription. Although RSK2 was reported to be activated by FLT3-ITD or upregulated through PIM and to be required for leukemogenesis by FLT3-ITD but not by BCR/ABL, these studies examined only activation of CTKD, used mainly the CTKD inhibitor FMK, and did not adequately examine the downstream signaling mechanisms from RSK. Thus, we examined the significance of RSK in FLT3-ITD AML and the underlying molecular mechanisms.
The RSK NTKD inhibitors LJI308 and LJH685 as well as FMK distinctively inhibited FLT3-ITD-dependent proliferation of the AML cell line MV4-11 and model hematopoietic cell lines, whereas BCR/ABL-dependent proliferation of various cells were resistant. Furthermore, inhibition of FLT3-ITD by quizartinib or gilteritinib, but not that of BCR/ABL by imatinib, prominently inhibited RSK1 and RSK2 NTKDs, as examined by the activation-specific phosphorylation in their T-loop sites, associated with inhibition of the MEK/Erk pathway and PDK1 in MV4-11. Inhibition of FLT3-ITD also reduced expression of RSK1 but not RSK2, which, however, was not mimicked by inhibition of PIM. Intriguingly, FMK or LJI308 inhibited FLT3-ITD and phosphorylation of its substrate STAT5 in MV4-11 cells as well as in primary AML cells expressing FLT3-ITD.
By using the most specific RSK inhibitor LJH685 as well as sh-RNA-mediated knock down, CRISPR/CAS9-mediated knockout, or overexpression of RSK1 or RSK2 in MV4-11 cells, we found that mainly RSK1 but not RSK2 phosphorylates not only S6RP on S235/236 but also TSC2 on S1798 to upregulate the mTORC1/S6K/4EBP1 pathway. RSK1 also phosphorylated eIF4B on S422 and, in cooperation with PIM, on S408, which is required for efficient cap-dependent translation by the eIF4F complex. Furthermore, RSK1 phosphorylated Bad on S112, which is instrumental for dissociation from anti-apoptotic proteins Bcl-2 and Bcl-xL. On the other hand, inhibition of RSK or Erk by LJH685 or SCH772984, respectively, increased the expression level of BimEL in the S65-phosphorylated or unphosphorylated form, respectively, which is consistent with the idea that RSK phosphorylates BimEL after its phosphorylation on S65 by Erk to induce proteasomal degradation of this potent pro-apoptotic protein. Interestingly, inhibition of RSK upregulated the MEK/Erk pathway, which correlated with increased binding of SHP2 with Gab2 phosphorylated by FLT3-ITD and was prevented by the SHP2 inhibitor IIB-08, thus revealing the negative feed back mechanisms involving the Gab2/SHP2 complex.
LJH685 or RSK1 knockout reduced viable cell numbers of MV4-11 as well as FLT3-ITD-positive primary AML cells synergistically with the PIM inhibitor AZD-1208 or the PI3K inhibitor GDC-0941 at least partly by inducing apoptosis, which was prevented by overexpression of Bcl-xL or Mcl-1. Knock down of mTOR in MV4-11 cells also enhanced apoptosis induced by inhibition of PIM or PI3K. Moreover, LJH685 sensitized MV4-11 significantly to BH3 mimetics, such as venetoclax or A-1210477, which inhibits Bcl-2 or Mcl-1, respectively.
Our findings reveal that FLT3-ITD but not BCR/ABL activates RSK1 as well as RSK2 and upregulates RSK1 expression to promote proliferation and survival of leukemic cells through upregulation of the mTORC1 pathway and modification of eIF4B in collaboration with the PI3K/Akt and STAT5/PIM pathways and additionally through inhibitory modification of the proapoptotic proteins Bad and Bim (Figure). Thus, RSK1 represents a promising molecular target particularly in combination with PIM or PI3K for novel therapeutic strategies against therapy-resistant FLT3-ITD-positive AML.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal