

Background:
Leukemia stem cells (LSCs) and acute myeloid leukemia (AML) blasts persisting in the bone marrow (BM) after chemotherapy are key drivers of AML relapse and chemotherapy refractoriness. The hypoxic microenvironment of the BM is known to protect these cells, however the mechanisms behind this chemoresistance are not well understood.
We have previously shown that AML cell lines cultured under hypoxia upregulate autophagy. Blocking autophagy with lysosome-based autophagy inhibitor Bafilomycin A1 (Baf A1) was shown to target LSCs in colony formation (CF) assays and in vivo serial transplants in NSG mice. Another autophagy inhibitor chloroquine (CQ) also demonstrated anti-LSC effects in CF assays. The clinical development of Baf A1 and CQ has been limited due to poor pharmacokinetics and toxicity. Lys05 is a promising CQ derivative with increased potency and therapeutic potential.
Recent studies have shown that LSCs have an increased reliance on oxidative phosphorylation (OXPHOS) as well as mitophagy, the autophagic degradation of damaged mitochondria. Hypoxia has also been demonstrated to increase mitophagy in certain cell types through upregulation of BNIP3. We hypothesized that AML cells cultured under hypoxia would have an increased reliance on mitophagy for mitochondrial homeostasis and survival. Treatment with autophagy inhibitors would be expected to block mitophagy and induce AML cell death specifically under hypoxia.
Methods:
Human AML cells (MOLM13) were cultured under hypoxia (1% O2) or normoxia (21% O2). NSG mice were inoculated with luciferase expressing MOLM13-BLIV & treated with vehicle (DMSO) or Lys05 (40 mg/kg) IP for 18 days. Tumor burden was assessed by bioluminescence. CF assays were established with AML patient cells in the presence of vehicle or Lys05 in MethoCult under normoxia/hypoxia and counted on day 13. Annexin V-FITC/PI flow cytometry was used to measure apoptosis. OXPHOS was assessed with a Seahorse XFe96 using the Mitochondrial Stress Test. Mitochondrial mass was measured by flow cytometry using MITO-ID Green. BNIP3 & PINK1 protein expression was visualized via western blot.
Results:
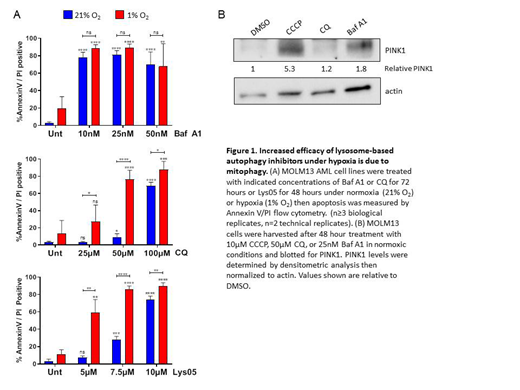
Treatment with Lys05 decreased in vivo tumor burden significantly in NSG mice systemically engrafted with human AML cells. We also saw a marked decrease in the number of CF-units in primary AML patient samples treated with Lys05 which was further enhanced under hypoxia. Treatment of AML cell lines with CQ and Lys05 also enhanced apoptosis under hypoxia as compared to normoxia. Baf A1, however, showed equal amounts of apoptosis in normoxia and hypoxia (Fig. 1A).
While Baf A1, CQ, and Lys05 all inhibit autophagy through deacidification of the lysosome, Baf A1 has been shown to induce additional effects on mitochondria, inducing uncoupling of OXPHOS and depolarization. We therefore examined the autophagy inhibitors' effect on mitochondrial function. At 24 hours, Baf A1 caused a significant decrease in basal and maximal respiration under both hypoxia and normoxia. CQ and Lys05, however, only showed a significant decrease in OXPHOS under hypoxia, with no effect on mitochondrial function under normoxia.
We postulated that this effect arose from increased mitophagy under hypoxia. BNIP3 expression levels were enhanced under hypoxia in MOLM13 cells at 24- 48 hours. If mitophagy is constitutively occurring, blocking this process would cause an increase in the number of mitochondria. As expected, the number of mitochondria increased when treated with Lys05 or CQ under hypoxia, but not under normoxia, suggesting that mitophagy is occurring only under hypoxic conditions. Baf A1 caused an increase in mitochondria under both hypoxia and normoxia, suggesting that Baf A1 can both induce mitophagy and block mitochondrial degradation. We further confirmed this was due to mitophagy by assessing expression of the mitophagy protein PINK1. In normoxia, Baf A1 showed an almost 2-fold increase in PINK1 compared to the vehicle whereas CQ did not (Fig. 1B).
Conclusion:
We have identified a class of autophagy inhibitors that displays enhanced efficacy in AML cells under hypoxic conditions that reflect the BM microenvironment. This is due in part to their ability to target AML cellular reliance on mitophagy. These results provide the rationale for the further clinical development of Lys05 or other lysosome-based autophagy inhibitors as a novel means of targeting minimal residual disease in AML therapy.
Guzman:Samus Therapeutics: Patents & Royalties: intellectual rights to the PU-FITC assay; SeqRx: Consultancy; Cellectis: Research Funding. Wang:Pfizer: Other: Advisory role, Speakers Bureau; Stemline: Other: Advisory role, Speakers Bureau; Daiichi: Other: Advisory role; Amgen: Other: Advisory role; Agios: Other: Advisory role; Abbvie: Other: Advisory role; Kite: Other: Advisory role; Jazz: Other: Advisory role; Astellas: Other: Advisory role, Speakers Bureau; celyad: Other: Advisory role.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal