

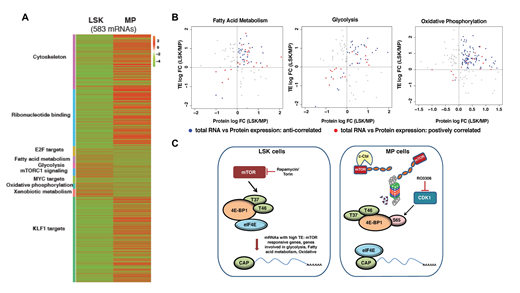
Prior studies in numerous biological systems have shown that alterations in mRNA expression frequently fail to predict changes in protein expression. This may be due to many regulatory mechanisms that occur post-transcriptionally including mRNA recruitment to ribosomes, translational initiation, ribosome processivity, and protein stability, among others. Indeed, several examples of selective translation of mRNAs has been described both in malignant and normal cells. To determine the extent and potential impact of translational reprogramming on early hematopoietic development, we performed an integrated analysis of total RNA, polysome RNA, and whole proteome data generated from HSC-enriched LSK (Lin-Sca-1+c-Kit+) and MP (Lin-Sca-1-c-Kit+) cells from mouse. Our studies revealed that although LSK cells show lower global translation than MPs, they exhibited significantly higher translational efficiency (TE = polysome/total RNA abundance) of mRNAs supporting processes required for HSC maintenance (e.g. glycolysis, fatty acid metabolism, oxidative phosphorylation, mTOR signaling) (Fig 1A). Additionally, integrated analysis of proteomic and RNA expression data showed that, TE changes better predicted protein expression changes for these pathways, than total RNA expression (Fig1B).
Biochemical characterization of MP cells revealed markedly decreased mTOR protein expression and signaling in MP cells, especially in GMP and MEP. This is mediated through proteasomal degradation of mTOR protein. An E3 ligase prediction algorithm, identified c-Cbl as a potential candidate, targeting mTOR, which was confirmed by demonstrating the aberrant expression of mTOR in MPs in c-Cbl KO mice. In vitro and in vivo mTOR inhibition studies confirm that the MPN-like phenotype of c-Cbl KO mice, is due to aberrant activation of mTOR signaling in committed myeloid progenitors. Intriguingly, despite decreased expression of mTOR protein in MP cells, 4E-BP1, a known target of mTOR, was still phosphorylated at Ser-65- a critical step for initiating cap-dependent translation. Through a combination of prediction algorithms and candidate gene experimental approaches, we show that the critical phosphorylation event at Ser-65 is mediated by , as immunoprecipitation studies show physical association between CDK1 and 4E-BP, and pharmacological inhibition of CDK1 activity, reduced 4E-BP P-Ser-65 levels.
Overall, our data provide the first comprehensive characterization of the translatome in early hematopoiesis and demonstrated that the LSK to MP transition is characterized by significant translational reprogramming. This is, in part, mediated by the activation of a unique, mTOR-independent pathway to activate cap-dependent translation through the concerted action of c-Cbl and CDK1 to induce degradation of mTOR and phosphorylate 4E-BP to activation translation, respectively. Abrogation of the downregulation of mTOR signaling in myeloid progenitors, results in expansions of numerous myeloid lineages including neutrophils, monocytes and platelets (Fig 1C). Thus, our studies demonstrate the importance of proper translational reprogramming in early hematopoiesis.
Figure legend. (A) Heatmap showing pathways significantly enriched in LSK and or MP cells based on TE. (B) Comparison of TE to protein expression in LSK cells for genes involved in the indicated biological processes (Blue dots: mRNAs that showed an anticorrelation between total RNA and protein expression; Red dots: mRNAs that showed a positive correlation between total RNA and protein expression). (C) Model for translational reprogramming in early hematopoiesis. Despite lower rates of global translation, LSK cells show preferential translation of mRNAs sensitive to mTOR inhibition and required for HSC maintenance. In contrast, in highly translating MP cells, loss of mTOR expression is mediated by the E3 ubiquitin ligase c-Cbl. When c-Cbl is deleted and mTOR protein is aberrantly expressed, this results in increased mature myeloid output. In the absence of mTOR, eIF4E-cap-dependent translation is maintained through the action of CDK1, which phosphorylates the S65 residue of 4E-BP1 to release eIF4E.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal