

Deficiency of the plasma protein factor XI (FXI) may be associated with excessive trauma-induced bleeding, particularly when injury involves the nasopharynx, mouth or urinary tract.1-3 In this issue of Blood, present data that point to a novel approach for treating or preventing bleeding in this disorder.4
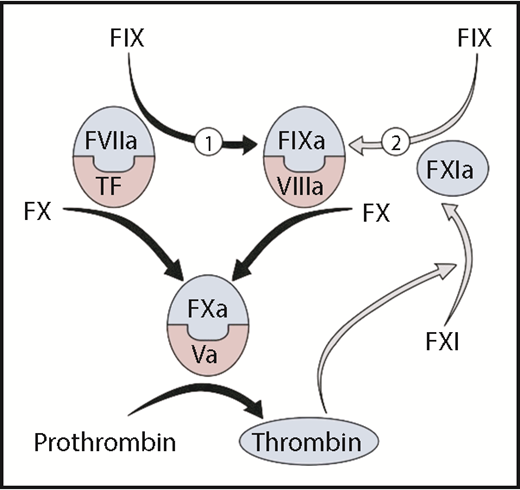
Thrombin generation is initiated when the protease factor VIIa and the cofactor tissue factor form a complex that converts factor X to factor Xa. Factor Xa, with its cofactor factor Va, catalyzes conversion of prothrombin to thrombin. Factor VIIa/tissue factor also converts factor IX to the protease factor IXa (reaction 1), which, in the presence of factor VIIIa, activates additional factor X to sustain thrombin generation. Factor IX is also converted to factor IXa by factor XIa (reaction 2). In the original cascade/waterfall models of coagulation, factor XI was activated by the enzyme factor XIIa during a process called contact activation. However, the absence of abnormal bleeding in factor XII deficient individuals indicates that other mechanisms exist for factor XI activation. In the figure, factor XI is activated by thrombin generated initially through the factor VIIa/tissue factor complex, creating a feedback loop (gray arrows) for factor IX activation. Coagulation protease precursors are indicated by black type, and the active forms of the proteases are shown within blue half-ovals. Cofactors are shown within pink half-ovals. F, factor.
Thrombin generation is initiated when the protease factor VIIa and the cofactor tissue factor form a complex that converts factor X to factor Xa. Factor Xa, with its cofactor factor Va, catalyzes conversion of prothrombin to thrombin. Factor VIIa/tissue factor also converts factor IX to the protease factor IXa (reaction 1), which, in the presence of factor VIIIa, activates additional factor X to sustain thrombin generation. Factor IX is also converted to factor IXa by factor XIa (reaction 2). In the original cascade/waterfall models of coagulation, factor XI was activated by the enzyme factor XIIa during a process called contact activation. However, the absence of abnormal bleeding in factor XII deficient individuals indicates that other mechanisms exist for factor XI activation. In the figure, factor XI is activated by thrombin generated initially through the factor VIIa/tissue factor complex, creating a feedback loop (gray arrows) for factor IX activation. Coagulation protease precursors are indicated by black type, and the active forms of the proteases are shown within blue half-ovals. Cofactors are shown within pink half-ovals. F, factor.
Factor XI is the precursor of factor XIa, a protease that contributes to coagulation primarily by converting factor IX to its active form factor IXa.5,6 In current schemes of thrombin generation, the main activator of factor IX at an injury site is a complex formed between the protease factor VIIa and the membrane-associated cofactor tissue factor (see figure, reaction 1). The importance of this reaction is reflected in the severe bleeding that accompanies deficiency of factor IX or its cofactor factor VIII (hemophilia B and A, respectively). For most injuries, factor IXa generated by factor VIIa/tissue factor appears to be sufficient for hemostasis. However, some situations may require factor XIa-mediated factor IX activation (see figure, reaction 2), perhaps to supplement factor IXa generated by factor VIIa/tissue factor or to sustain factor IX activation. It has been proposed that the predilection of patients with factor XI deficiency to bleed after injury to the mouth, nose or urinary tract reflects a requirement for additional factor IX activation to counter robust fibrinolytic activity intrinsic to these areas of the body.1-3
Replacement therapy in factor XI deficient patients presents several challenges. Plasma contains relatively small amounts of the protein, and large volumes may be required to significantly change the plasma concentration.2,3 Factor XI concentrates are not widely available, and their use has been limited by concerns that they trigger thrombotic events in some patients.7 Additionally, about one-third of patients completely lacking factor XI develop neutralizing alloantibody inhibitors after replacement therapy, comprising subsequent treatment.8 Recombinant factor VIIa has been used successfully to address the limitations of replacement therapy in factor XI deficiency,2,3 suggesting that enhanced factor VIIa/tissue factor activity compensates for the absence of factor XI.
By using thrombin generation assays, Bakhtiari and Meijers observed that supplementing factor XI deficient plasma with the recombinant factor IX concentrate Benefix enhanced thrombin formation initiated by a low concentration of tissue factor.4 The effect persisted after the addition of a neutralizing antibody to factor XI, which indicates that trace contamination of the system with factor XI or XIa was not responsible. Furthermore, a neutralizing antibody to factor IX blocked the contribution of Benefix, which argues against a contribution from an unsuspected non-factor IX contaminant. Surprisingly, thrombin generation was not enhanced by a concentrate of plasma-derived factor IX (Nonafact). This pointed to a specific property of Benefix as the enhancer of thrombin generation. Benefix, it turns out, contains a small amount (∼0.07%) of factor IXa, whereas Nonafact does not. The factor IXa may bypass the blockade created by factor XI deficiency in the feedback loop indicated by the gray arrows in the figure.
These intriguing findings raise the prospect that infusion of some currently available factor IX concentrates may be useful for treating bleeding episodes or for preventing bleeding prior to invasive procedures in factor XI–deficient patients. There is substantial safety data on hand for these preparations, and they are widely available. What must be demonstrated is the clinical efficacy of such an approach. Thrombin generation assays can be quite sensitive to trace amounts of coagulation proteases, and it is not a given that the impressive effects observed in vitro will translate to improved hemostasis in vivo. The results with Benefix and Nonafact demonstrate that factor IXa levels vary between factor IX concentrates, and the most appropriate preparations need to be identified. Furthermore, because we do not know why factor IXa is present in some factor IX preparations and not others, it is conceivable that there could be substantial variability in the protease level between lots of the same product. Demonstrating therapeutic benefit of a treatment in factor XI deficient patients is difficult because of the highly variable nature of the bleeding disorder between patients, and even in the same patient at different times. Variations in the amount of factor IXa in a factor IX concentrate would make assessment of treatment efficacy even more challenging. Although much work remains to be done, the observations of Bakhtiari and Meijers suggest an addition to our therapeutic armamentarium for factor XI deficiency. This would be most welcome, particularly in areas of the world where factor XI concentrates are not easily obtained.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal