

Key Points
Biallelic EFL1 mutations cause Shwachman-Diamond syndrome.
EFL1 deficiency impairs eIF6 eviction from nascent 60S ribosomal subunits, reducing subunit joining and attenuating protein synthesis.

Abstract
Shwachman-Diamond syndrome (SDS) is a recessive disorder typified by bone marrow failure and predisposition to hematological malignancies. SDS is predominantly caused by deficiency of the allosteric regulator Shwachman-Bodian-Diamond syndrome that cooperates with elongation factor-like GTPase 1 (EFL1) to catalyze release of the ribosome antiassociation factor eIF6 and activate translation. Here, we report biallelic mutations in EFL1 in 3 unrelated individuals with clinical features of SDS. Cellular defects in these individuals include impaired ribosomal subunit joining and attenuated global protein translation as a consequence of defective eIF6 eviction. In mice, Efl1 deficiency recapitulates key aspects of the SDS phenotype. By identifying biallelic EFL1 mutations in SDS, we define this leukemia predisposition disorder as a ribosomopathy that is caused by corruption of a fundamental, conserved mechanism, which licenses entry of the large ribosomal subunit into translation.
Introduction
Ribosomes are the essential macromolecular machines that carry out protein synthesis in all organisms. Eukaryotic ribosomes are formed by the association of the small 40S subunit (containing the 18S ribosomal RNA [rRNA]) and the large 60S subunit (containing the 28S, 5.8S, and 5S rRNAs) to form actively translating 80S ribosomes.1 In humans, mutations in the ribosome assembly factors or ribosomal proteins themselves lead to disorders termed ribosomopathies2 that are associated with a range of clinical phenotypes, including bone marrow failure, hematological malignancies, and cancer.2-4
Shwachman-Diamond syndrome (SDS) is an autosomal recessive disorder associated with bone marrow failure and an increased risk of progression to hematological malignancies, including myelodysplastic syndrome (MDS) and acute myeloid leukemia.5 SDS is also characterized by developmental anomalies, including poor growth, exocrine pancreatic dysfunction, metaphyseal chondrodysplasia, and cognitive impairment.6,7 In ∼90% of cases, SDS is caused by biallelic mutations in the SBDS gene. Genetic, biochemical, and structural studies have revealed that the Shwachman-Bodian-Diamond syndrome (SBDS) protein cooperates with elongation factor-like GTPase 1 (EFL1), a homolog of the ribosomal translocase elongation factor 2 (EF-2), during the terminal step in 60S subunit maturation to couple release of the ribosome antiassociation factor eIF6 (yeast Tif6) to a final quality-control assessment of the integrity of the P-site and the GTPase center of the 60S subunit.4 By binding to the sarcin-ricin loop and ribosomal proteins uL3, uL14, and eL24 (using the new nomenclature8 ) on the intersubunit face of the 60S subunit, eIF6 forms a physical barrier to ribosomal subunit joining4,9,10 and must therefore be removed to activate translation. However, an alternative model posits that protein kinase C (PKC) and the receptor for activated protein C (RACK1) promote eIF6 eviction through phosphorylation of eIF6 on residue S235.11
Approximately 10% of individuals with SDS lack mutations in the SBDS gene. The identification of biallelic mutations in DNAJC21 (yeast Jjj1) in a subset of SDS patients lacking mutations in the SBDS gene12-15 indicates that SDS is genetically heterogeneous. Jjj1 cooperates with the cytoplasmic zinc-finger protein Rei1 (human ZNF622) to stimulate the ATPase activity of the Hsp70 chaperone Ssa (human HSPA8), liberating the nuclear export factor Arx1 (human PA2G4) from its binding site near the peptide exit tunnel on the 60S subunit.16-20 These data raise the possibility that additional components of the 60S subunit assembly pathway might also be mutated in SDS.
Here, we report the identification of biallelic mutations in the EFL1 gene in individuals with clinical features of SDS. We demonstrate that disease-associated EFL1 mutations disrupt eIF6 release from late cytoplasmic 60S subunits, thereby impairing ribosomal subunit joining and attenuating global translation. Furthermore, we show that Efl1-mutant mice recapitulate key aspects of the SDS phenotype. We define SDS as a ribosomopathy that is caused by corruption of a fundamental, conserved mechanism, which licenses the entry of the large ribosomal subunit into translation.
Materials and methods
Patients
Informed consent was obtained from the families in accordance with the Declaration of Helsinki. This study was approved by the institutional review board of INSERM and Assistance Publique–Hôpitaux de Paris.
Cell culture
Detailed procedures are described in supplemental Materials and methods (available on the Blood Web site).
Immunoblotting
Immunoblotting procedure is detailed in detailed in supplemental Materials and methods. Antibodies are listed in supplemental Table 3.
Whole-exome sequencing
The whole-exome sequencing procedure is detailed in supplemental Materials and methods.
Targeted resequencing by NGS. Detailed procedure is described in supplemental Materials and methods.
cDNA analysis
Information on complementary DNA (cDNA) analysis is provided in supplemental Materials and methods.
Constructs
The EFL1 open reading frame was polymerase chain reaction amplified and cloned into the lentiviral vectors p.lenti7.3/V5 TOPO (Invitrogen) and pCW57-GFP-P2A (Plasmid #71783; Addgene) for complementation experiments.
Measurement of protein synthesis
Measurement of protein synthesis, detailed in supplemental Materials and methods, was performed as described previously.21
Sucrose density gradients
Cell fractionation
Detailed procedures are described in supplemental Materials and methods.
eIF6 release assay
Detailed information is provided in supplemental Materials and methods.
EFL1 cloning, expression, and purification
Human EFL1 mutants were generated by polymerase chain reaction using the wild-type EFL1 cDNA (long isoform ENST00000268206.11). Detailed procedures are available in supplemental Materials and methods.
Statistics
The Student t test (2 tailed, type 2) was used to determine significant differences. P < .05 was considered significant.
Mice
Detailed procedures are described in supplemental Materials and methods.
Results
Identification of biallelic EFL1 mutations in 3 unrelated individuals
In the course of a systematic survey of individuals presenting with inherited bone marrow failure syndrome, we performed whole-exome sequencing analysis to identify rare biallelic variants predicted to be pathogenic (frequency <0.1% in 1000 Genomes, the Exome Variant Server, the Single Nucleotide Polymorphism Database, and our in-house database [8319 individuals]). This approach identified a male (P1) carrying compound heterozygous mutations in the EFL1 gene (HUGO Gene Nomenclature Committee, 25,789; Entrez Gene, 79,631) encoding EFL1 (Figure 1A). Since EFL1 physically and functionally interacts with SBDS,4,22-24 we considered EFL1 as a strong candidate gene. Sanger sequencing confirmed the presence of biallelic EFL1 mutations, including a nonsense mutation causing a premature stop codon on 1 allele (c.2260C>T; p.R754*) inherited from his healthy mother and also present in his healthy sister (Figure 1A; supplemental Figure 1A) and a missense mutation leading to an amino acid substitution (c.1514T>C; p.F505S) on the allele inherited from his unaffected father (Figure 1A; supplemental Figure 1A). EFL1 cDNA sequencing in P1 suggested reduced abundance of the allele carrying the c.2260C>T; p.R754* mutation, consistent with partial nonsense-mediated decay (supplemental Figure 1A).
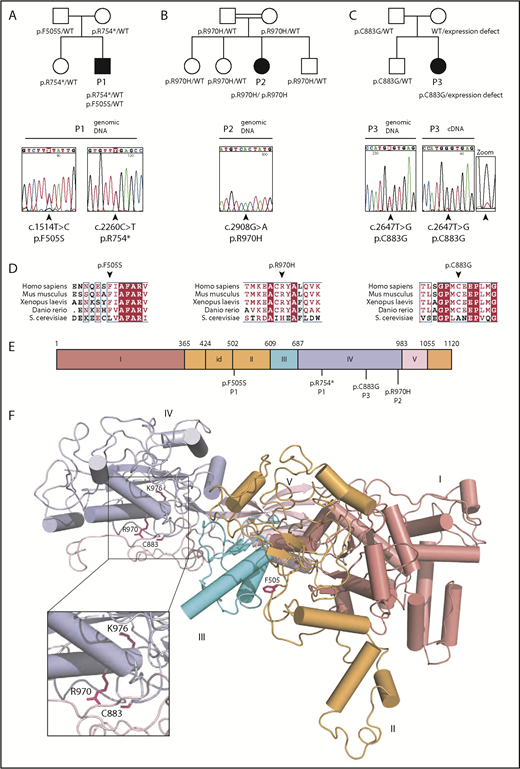
Identification of EFL1 mutations in 3 individuals with SDS. Family pedigrees and direct Sanger sequencing of the EFL1 gene in individuals 1 (P1) (A), 2 (P2) (B), and 3 (P3) (C). Arrows indicate the position of the mutations. (D) Multiple sequence alignment of EFL1 proteins from representative species. Identical amino acids are shown in red with white characters, similar amino acids are in red character, and a blue frame represents similarity across groups. (E) Schematic of the domain architecture of human EFL1 showing the position of disease-associated mutations. Domains I to V are indicated; id, insertion domain (residues 424-502) that distinguishes EFL1 from other translational GTPases. (F) Mapping of SDS-associated mutations onto human EFL1 (Protein Data Bank accession number 5anc4 ), shown in ribbon representation. Domains are colored deep salmon (I), light orange (id, II), cyan (III), and light blue (IV). Residue K976 corresponds to K983 mutated in the N-ethyl-N-nitrosourea mutant mouse model (see below). WT, wild-type.
Identification of EFL1 mutations in 3 individuals with SDS. Family pedigrees and direct Sanger sequencing of the EFL1 gene in individuals 1 (P1) (A), 2 (P2) (B), and 3 (P3) (C). Arrows indicate the position of the mutations. (D) Multiple sequence alignment of EFL1 proteins from representative species. Identical amino acids are shown in red with white characters, similar amino acids are in red character, and a blue frame represents similarity across groups. (E) Schematic of the domain architecture of human EFL1 showing the position of disease-associated mutations. Domains I to V are indicated; id, insertion domain (residues 424-502) that distinguishes EFL1 from other translational GTPases. (F) Mapping of SDS-associated mutations onto human EFL1 (Protein Data Bank accession number 5anc4 ), shown in ribbon representation. Domains are colored deep salmon (I), light orange (id, II), cyan (III), and light blue (IV). Residue K976 corresponds to K983 mutated in the N-ethyl-N-nitrosourea mutant mouse model (see below). WT, wild-type.
In order to identify additional individuals potentially carrying biallelic EFL1 mutations, we analyzed the EFL1 gene by gene-targeted sequencing of a cohort of 30 individuals who were diagnosed clinically with SDS but lacked mutations in the SBDS gene. This approach identified an EFL1 homozygous missense mutation (c.2908C>T; p.R970H) in a female (P2), born from a consanguineous family (Figure 1B). Sanger sequencing confirmed the mutation and indicated that both parents and the healthy siblings were heterozygous for the mutant EFL1 allele (Figure 1B; supplemental Figure 1B). In addition, we identified a third female (P3) carrying a heterozygous EFL1 missense mutation (c.2647T>G; p.C883G) (Figure 1C). Sanger sequencing confirmed the mutation and indicated that it was inherited from her father and also carried by her healthy brother (Figure 1C; supplemental Figure 1C). No other pathogenic exon-encoded variants were detected in individual P3. However, the near absence of detectable wild-type EFL1 messenger RNA transcripts from the “normal” allele in P3’s cells suggests a noncoding defect in the allele inherited from her mother that affects EFL1 expression (Figure 1C, zoom). Sequence analysis of genomic DNA and cDNA of a synonymous single-nucleotide variant present in a heterozygous state in the EFL1 coding sequence (rs4725) from a healthy control, P3, and P3’s mother confirmed the defective expression of 1 EFL1 allele in both P3 and her mother (supplemental Figure 2A). Furthermore, immunoblotting analysis demonstrated reduced EFL1 protein expression in B lymphoblastoid cell line (B-LCL) lysates from the mother of P3 compared with control, confirming the genetic results (supplemental Figure 2B). Haplotype analysis of the EFL1 locus in P3’s family indicated that P3’s brother, unlike his sister, inherited the wild-type maternal EFL1 allele (supplemental Figure 2C). However, whole EFL1 gene sequencing did not identify obvious genetic variants that could explain the defective expression of EFL1 from the maternal allele. Nonetheless, biochemical analysis confirmed a functional EFL1 defect in individual P3 (see below).
The 4 identified EFL1 mutations affected both the long (ENST00000268206.11) and short (ENST00000359445.7) EFL1 isoforms. Furthermore, they were absent or extremely rare in the gnomAD Browser (supplemental Table 1) and in silico tools (sorting intolerant from tolerant,25 PolyPhen-2,26 and combined annotation-dependent depletion27 ) and the American College of Medical Genetics and Genomics standards28 predicted the EFL1 coding variants to be deleterious (supplemental Table 1). Moreover, the 3 missense mutations affect highly conserved amino acids across species (Figure 1D). Interestingly, the EFL1 p.F505S mutation maps to a key functional dynamic interface between EFL1 domains II and III (Figure 1E-F) that mediates EFL1 conformational change on the ribosome.4 In yeast, mutations that map to the EFL1 domain II and III interface likely suppress mutations in the P-site loop of uL16 (RPL10) by promoting conformational changes similar to those undergone by eukaryotic EF-2 and EF-G during translocation.29 Both the p.C883G and p.R970H missense mutations map to the interface between EFL1 domain IV (Figure 1E-F) and the C-terminal domain III of SBDS (Figure 2). In yeast, domain IV is essential for Efl1 function in vivo.29 Based on these observations, we hypothesized that the p.F505S, p.R970H and p.C883G mutations impair EFL1 function and that there is a causal link between the EFL1 mutations identified and the SDS phenotype.
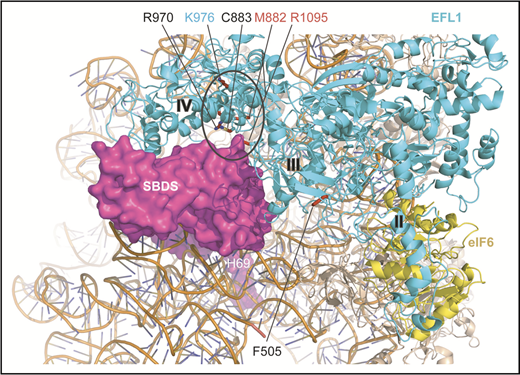
SDS-associated EFL1 mutations map to key functional interfaces. SBDS C-terminal domain, magenta, space-filling representation; EFL1, cyan; eIF6, yellow (Protein Data Bank accession number 5anb), ribbon representation. EFL1 domains II, III, and IV are indicated. EFL1 residues targeted by mutations that map to the interface with SBDS are encircled. Residues mutated in SDS are indicated in black (this study) or red (Stepensky et al41 ) text; residue K976 (cyan) is targeted in the mouse model (this study). H69, rRNA helix 69.
SDS-associated EFL1 mutations map to key functional interfaces. SBDS C-terminal domain, magenta, space-filling representation; EFL1, cyan; eIF6, yellow (Protein Data Bank accession number 5anb), ribbon representation. EFL1 domains II, III, and IV are indicated. EFL1 residues targeted by mutations that map to the interface with SBDS are encircled. Residues mutated in SDS are indicated in black (this study) or red (Stepensky et al41 ) text; residue K976 (cyan) is targeted in the mouse model (this study). H69, rRNA helix 69.
Clinical features of EFL1-deficient individuals
P1 is a male from a French family (Table 1) who was born at term with a birth weight of 2480 g (<3.0 centile) and a height of 47 cm (3.0 centile). He initially presented with unsteady gait. He sustained a spontaneous femoral fracture at 5 years of age and required surgical correction of severe genu valgum at 12 years of age. Metaphyseal chondrodysplasia was noted on a radiograph at 13 years of age (supplemental Figure 3A). At 17 years of age, he developed mild thrombocytopenia and subsequently progressive leukopenia with neutropenia and mild anemia (supplemental Figure 3B). At 27 years of age, he developed chronic diarrhea, with reduced fat-soluble vitamins and fecal elastase, consistent with exocrine pancreatic dysfunction. He developed portal hypertension and splenomegaly secondary to liver cirrhosis. Despite mild intellectual impairment, he was able to perform daily work. At 31 years of age, the bone marrow trephine was hypocellular, with typical hyposegmented neutrophils observed in SDS5,30 (Table 1; supplemental Figure 3C). No clonal cytogenetic abnormality was detected.
Individuals with EFL1 mutations meet the clinical criteria for SDS
| Patient P1 | Patient P2 | Patient P3 | |
|---|---|---|---|
| Sex | M | F | F |
| Geographic origin | France | Africa Guinea | France |
| Consanguinity | No | Yes | No |
| Birth features | At term | At term | At term |
| Weight | 2840 (<3 centile) | 2640 g (2.5 centile) | 2100 g (0.1 centile) |
| Height | 47 cm (3 centile) | 46 cm (1.7 centile) | 45 cm (1.2 centile) |
| Head circumference | NA | 32.5 cm (6.8 centile) | NA |
| Evidence of bone marrow failure | |||
| Initial manifestation and age at presentation | Thrombocytopenia, age 17 y | Failure to thrive, age 1.5 y | Anemia, failure to thrive age, 1.2 mo |
| Neutropenia | Mild | Severe | Mild |
| Absolute neutrophil count (×109/L), median (min-max) | 1.05 (1.0-3.9) | 8.2 (1.0-37) | 2.48 (1.83-4.0) |
| G-CSF | No | No | No |
| Red cell transfusion | No | No | Yes, during first year |
| Hemoglobin (g/L), median (min-max) | 118 (75-144) | 104 (85-137) | 114 (74−182) |
| Thrombocytopenia | Severe | Mild | During follow up, thrombocytopenia (51 × 109/L) was observed only once, without hemorrhagic symptoms; count recovered spontaneously |
| Platelets (×109/L), median (min-max) | 32 (13-168) | 110 (47-231) | 201 (51-326) |
| Reticulocytes (×109/L) | 109 | 72 | 73 |
| MCV (fL) | 90 | 94 | 103 |
| HbF (%) | NA | 5.8 (age 8 y) | NA |
| Platelet transfusion | No | No | No |
| Pancytopenia | Yes, after age 30 y | No | No |
| Bone marrow cellularity | Low (<5%); 2 bone marrows (at age 28 y) reviewed; no myeloid maturation arrest; segmentation defect in 8-22% of mature neutrophils | Low; at age 8 y, no myeloid maturation arrest; segmentation defect in 35% of mature neutrophils | Normal in the first year of life, mildly decreased on the 3 bone marrow smears >1 y old; erythroblastopenia age 4 mo; no myeloid maturation arrest age 5 y, no erythroblastopenia; segmentation defect in 8% of mature neutrophils |
| Bone marrow cytogenetics | Normal | Normal | Normal |
| Evidence of pancreatic dysfunction | Yes, age 27 y | Yes, age 5 y | Yes, age 6 mo |
| Diagnosis of pancreatic insufficiency | Yes | Yes | Yes |
| Pancreatic enzyme therapy | Yes | Yes | Yes |
| Pancreatic MRI | Lipomatosis | No lipomatosis | No MRI, but hyperechogenicity on US |
| Chronic diarrhea | Yes | Yes | Yes |
| Fecal elastase | Low (<200 μg/g feces) | Low (<200 μg/g feces) | Very low (<100 μg/g feces) |
| Vitamin A | Normal | Low | Low |
| Vitamin D | Normal | Normal | Normal |
| Vitamin E | NA | Normal | Normal |
| Liver | Portal hypertension with splenomegaly. Fibrosis evaluated on CT scan and liver biopsy age 29 y; virology negative | Normal liver MRI; elevated liver enzymes AST/ALT (50-170 IU/L), then spontaneous recovery; no biopsy | Normal liver MRI; elevated liver enzymes AST/ALT (60-120 IU/L); no biopsy |
| Skeletal abnormalities | Claudication and atraumatic femoral neck fracture at 5 y of age; severe osteoporosis, polyepiphyseal and metaphyseal dysplasia with major bilateral genu valgum, patellar luxation, hip subluxation and bilateral oval femoral heads; surgery for genu valgum complicated by osteitis, but outcome was favorable; coxa plana and metaphyseal chondrodysplasia noted at age 29 y | No abnormalities | Metaphyseal dysplasia; skeletal defects of the vertebrae, limbs and ribs; pectus carinatum |
| Bone density | Low, T score (vertebrae) −3.2 SD | Not evaluated | Not evaluated |
| Growth impairment | Short stature −2.6 SD; no therapy | Short stature −3 SD (not improved by pancreatic enzyme replacement); GH deficiency; GH therapy | Short stature −6 SD (not improved by pancreatic enzyme replacement); GH deficiency; no therapy |
| Eyes | Normal | Very severe myopia with low visual acuity | Normal |
| Ears | Normal | Hypoacusia | Normal |
| Cognitive impairment and developmental delay | Normal | Mild; 2-y delay at a special school for learning disabilities; speech delay, but at age 10 y, WISC-IV test result1 normal for age | Mild; 3-y delay in special school for learning disabilities |
| Neurological symptoms | None | Ataxia; nystagmus; CNS MRI normal, but Chiari type I malformation diagnosed at 2 y | None; CNS MRI normal |
| Dentition | Abnormal tooth enamel | Numerous dental caries | Normal |
| Skin abnormalities | No | No | No |
| Recurrent bacterial Infections | No | No | Yes |
| Age at last follow up | 31 y | 14 y | 8 y |
| Vital status | Alive | Alive | Alive |
| Patient P1 | Patient P2 | Patient P3 | |
|---|---|---|---|
| Sex | M | F | F |
| Geographic origin | France | Africa Guinea | France |
| Consanguinity | No | Yes | No |
| Birth features | At term | At term | At term |
| Weight | 2840 (<3 centile) | 2640 g (2.5 centile) | 2100 g (0.1 centile) |
| Height | 47 cm (3 centile) | 46 cm (1.7 centile) | 45 cm (1.2 centile) |
| Head circumference | NA | 32.5 cm (6.8 centile) | NA |
| Evidence of bone marrow failure | |||
| Initial manifestation and age at presentation | Thrombocytopenia, age 17 y | Failure to thrive, age 1.5 y | Anemia, failure to thrive age, 1.2 mo |
| Neutropenia | Mild | Severe | Mild |
| Absolute neutrophil count (×109/L), median (min-max) | 1.05 (1.0-3.9) | 8.2 (1.0-37) | 2.48 (1.83-4.0) |
| G-CSF | No | No | No |
| Red cell transfusion | No | No | Yes, during first year |
| Hemoglobin (g/L), median (min-max) | 118 (75-144) | 104 (85-137) | 114 (74−182) |
| Thrombocytopenia | Severe | Mild | During follow up, thrombocytopenia (51 × 109/L) was observed only once, without hemorrhagic symptoms; count recovered spontaneously |
| Platelets (×109/L), median (min-max) | 32 (13-168) | 110 (47-231) | 201 (51-326) |
| Reticulocytes (×109/L) | 109 | 72 | 73 |
| MCV (fL) | 90 | 94 | 103 |
| HbF (%) | NA | 5.8 (age 8 y) | NA |
| Platelet transfusion | No | No | No |
| Pancytopenia | Yes, after age 30 y | No | No |
| Bone marrow cellularity | Low (<5%); 2 bone marrows (at age 28 y) reviewed; no myeloid maturation arrest; segmentation defect in 8-22% of mature neutrophils | Low; at age 8 y, no myeloid maturation arrest; segmentation defect in 35% of mature neutrophils | Normal in the first year of life, mildly decreased on the 3 bone marrow smears >1 y old; erythroblastopenia age 4 mo; no myeloid maturation arrest age 5 y, no erythroblastopenia; segmentation defect in 8% of mature neutrophils |
| Bone marrow cytogenetics | Normal | Normal | Normal |
| Evidence of pancreatic dysfunction | Yes, age 27 y | Yes, age 5 y | Yes, age 6 mo |
| Diagnosis of pancreatic insufficiency | Yes | Yes | Yes |
| Pancreatic enzyme therapy | Yes | Yes | Yes |
| Pancreatic MRI | Lipomatosis | No lipomatosis | No MRI, but hyperechogenicity on US |
| Chronic diarrhea | Yes | Yes | Yes |
| Fecal elastase | Low (<200 μg/g feces) | Low (<200 μg/g feces) | Very low (<100 μg/g feces) |
| Vitamin A | Normal | Low | Low |
| Vitamin D | Normal | Normal | Normal |
| Vitamin E | NA | Normal | Normal |
| Liver | Portal hypertension with splenomegaly. Fibrosis evaluated on CT scan and liver biopsy age 29 y; virology negative | Normal liver MRI; elevated liver enzymes AST/ALT (50-170 IU/L), then spontaneous recovery; no biopsy | Normal liver MRI; elevated liver enzymes AST/ALT (60-120 IU/L); no biopsy |
| Skeletal abnormalities | Claudication and atraumatic femoral neck fracture at 5 y of age; severe osteoporosis, polyepiphyseal and metaphyseal dysplasia with major bilateral genu valgum, patellar luxation, hip subluxation and bilateral oval femoral heads; surgery for genu valgum complicated by osteitis, but outcome was favorable; coxa plana and metaphyseal chondrodysplasia noted at age 29 y | No abnormalities | Metaphyseal dysplasia; skeletal defects of the vertebrae, limbs and ribs; pectus carinatum |
| Bone density | Low, T score (vertebrae) −3.2 SD | Not evaluated | Not evaluated |
| Growth impairment | Short stature −2.6 SD; no therapy | Short stature −3 SD (not improved by pancreatic enzyme replacement); GH deficiency; GH therapy | Short stature −6 SD (not improved by pancreatic enzyme replacement); GH deficiency; no therapy |
| Eyes | Normal | Very severe myopia with low visual acuity | Normal |
| Ears | Normal | Hypoacusia | Normal |
| Cognitive impairment and developmental delay | Normal | Mild; 2-y delay at a special school for learning disabilities; speech delay, but at age 10 y, WISC-IV test result1 normal for age | Mild; 3-y delay in special school for learning disabilities |
| Neurological symptoms | None | Ataxia; nystagmus; CNS MRI normal, but Chiari type I malformation diagnosed at 2 y | None; CNS MRI normal |
| Dentition | Abnormal tooth enamel | Numerous dental caries | Normal |
| Skin abnormalities | No | No | No |
| Recurrent bacterial Infections | No | No | Yes |
| Age at last follow up | 31 y | 14 y | 8 y |
| Vital status | Alive | Alive | Alive |
ALT, alanine aminotransferase; AST, aspartate amino transferase; CNS, central nervous system; G-CSF, granulocyte colony-stimulating factor; GH, growth hormone; HbF, hemoglobin F; IU, international unit; MCV, red blood cell mean corpuscular volume; MRI, magnetic resonance imaging; NA, not available; SD, standard deviation; U/K, unknown; WISC-IV, Wechsler Intelligence Scale for Children, Fourth Edition.
P2, a girl, is the third child in a consanguineous family of 4 (Table 1). She was born at 40 weeks of gestation with a birth weight of 2640 g (2.5 centile), a height of 46 cm (1.7 centile), and a head circumference of 32.5 cm (6.8 centile). She presented at 13 months of age with failure to thrive, anorexia, moderately elevated liver enzymes, reduced zinc and vitamin A levels, and mild iron deficiency anemia (Table 1; supplemental Figure 3B). Although antigliadin antibodies were positive at 8 months of age, they were later negative. At 18 months of age, ataxia and nystagmus were observed. She developed mild iron deficiency anemia (corrected with oral iron supplementation) and mild thrombocytopenia. At 3.8 years of age, she was neutropenic and thrombocytopenic (supplemental Figure 3B). In addition, exocrine pancreatic deficiency was noted, based on low fecal elastase and fat-soluble vitamins (A and E). An abdominal computed tomography scan showed no evidence of pancreatic lipomatosis. Bone marrow examination revealed hyposegmented neutrophils (Table 1; supplemental Figure 3C). She failed to achieve developmental milestones. Her visual acuity was very low (2/10 in both eyes) due to severe myopia (with no associated retinal abnormalities). She had normal social behavior with a normal Wechsler Intelligence Scale for Children, Fourth Edition test result31 at 10 years of age. However, her academic progress was delayed by 2 years at a special school for the blind. Although skeletal X-rays were normal, she exhibited growth delay due to deficiency of growth hormone, which was introduced at the age of 11 years. From 8 to 11 years of age, her growth rate decreased from −1.8 SD to −3 SD (height 125 cm) when growth hormone therapy was initiated. Two years later, her height was 138 cm (−2.7 SD), but puberty had not started.
P3, a girl, is a second child from a nonconsanguineous French family (Table 1). The older child and the parents are healthy. Growth delay and femoral shortening was noticed in utero at 32 weeks of gestation. Following a caesarean section at 39 weeks gestational age due to an abnormal cardiac rhythm, her birth weight was 2160 g (0.1 centile), height was 45 cm (1.2 centile), and pectus carinatum and short limbs were noted. Severe neonatal anemia and thrombocytopenia with abnormal liver function test results (3 × normal value) were observed at day 2. Transfusion-dependent anemia persisted until 2 years of age (supplemental Figure 3B) with erythroblastopenia and hyposegmented neutrophils in the bone marrow (Table 1; supplemental Figure 3C). Pancreatic insufficiency was diagnosed at 3 months of age, with chronic diarrhea, reduced fecal Elastase, and fat-soluble vitamins (A and E). Due to persistent anorexia, a gastrostomy was performed at 8 months of age and is still present at 5 years of age. At 2 years of age, she became red cell transfusion independent. Growth retardation persisted with a height of 88 cm (−6 SD) at age 7 years. She has major skeletal defects involving the vertebrae, limbs, and ribs, including metaphyseal chondrodysplasia (supplemental Figure 3A). She has good social skills but impaired cognitive development and is currently attending a special school with a 3-year delay.
In summary, all 3 individuals (P1, P2, and P3) exhibited hematological abnormalities, short stature, and exocrine pancreatic insufficiency consistent with the clinical diagnosis of SDS. However, all 3 individuals were negative for mutations in the SBDS gene.
Impaired eIF6 release and attenuated translation in cells from EFL1-mutant individuals
EFL1 cooperates with its cofactor SBDS to evict eIF6 from the nascent 60S ribosomal subunit during the terminal step in cytoplasmic maturation.4 We therefore set out to obtain in vivo evidence for impaired eIF6 recycling, defective ribosomal subunit joining, and impaired global protein translation in cells from individuals carrying EFL1 mutations. Immunoblotting revealed that EFL1 protein in cell lysates from P1 and P3’s fibroblasts was reduced to 15% and 5%, respectively, of wild-type levels (Figure 3A). Thus, biallelic EFL1 mutations in individual P1 reduced the expression and/or stability of the encoded EFL1 protein. Importantly, the low abundance of EFL1 in P3’s fibroblast lysates confirmed the sequencing results obtained from P3-derived EFL1 cDNA (Figure 1A, zoom; supplemental Figure 2). In contrast, the expression of EFL1 was not affected by the p.R970H mutation present in P2’s fibroblasts (Figure 3A). Similar findings were made in B-LCL lysates from the same individuals (Figure 3B). Furthermore, the expression of additional factors involved in terminal cytoplasmic ribosomal maturation (NMD3, DNAJC21, PA2G4, eIF6, and SBDS) was unaffected in EFL1-deficient cells (Figure 3A-B).
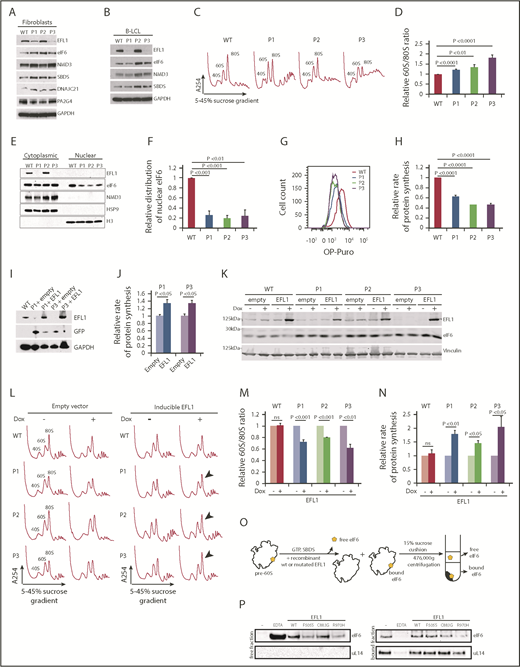
SDS-associated EFL1 mutations impair eIF6 release. (A) Total cell lysates from fibroblasts and (B) B-LCL from wild-type and 3 individuals (P1, P2, and P3) with SDS were immunoblotted to visualize the indicated proteins. (C) Defective ribosome assembly in EFL1 mutant fibroblasts. Polysome profiles from fibroblast extracts from 3 unrelated individuals with SDS compared with wild-type control. Quantification of the 60S:80S ribosomal subunit ratios is indicated as a bar chart (n ≥ 5) (D). EFL1 genotypes are provided in supplemental Table 1. (E) EFL1 is required for eIF6 recycling in human cells. Indicated proteins were visualized by immunoblotting in cytoplasmic or nuclear fractions from wild-type and EFL1 mutant fibroblasts. Histone H3, nuclear marker; HSP9, cytoplasmic marker. (F) Relative amount of eIF6 in the nucleus of EFL1 mutant cells compared with wild-type (n = 5). (G-H) EFL1 deficiency attenuates protein synthesis. OP-Puro incorporation in fibroblast cells lines from individuals P1, P2, and P3 relative to wild-type control cells quantified 1 hour after OP-Puro administration (n = 6). (I) Complementation of EFL1-deficient fibroblasts with wild-type EFL1. Lysates from EFL1 mutant fibroblasts transduced with a vector expressing GFP alone (+empty) or GFP + EFL1 (+EFL1) and from wild-type fibroblasts as a control. The indicated proteins were visualized by immunoblotting. (J) Wild-type EFL1 rescues global translation in patient fibroblast cell lines from P1 and P3 (n = 4). (K) Complementation of EFL1-deficient fibroblasts with inducible vector allows wild-type EFL1 expression after doxycycline (Dox) treatment. Lysates from EFL1 mutant fibroblasts transduced with an empty vector or EFL1-expressing vector with (+) or without (−) doxycycline. Vinculin is used as a loading control. (L) Comparison of polysome profiles from wild-type and EFL1-mutant fibroblasts transduced with empty vector or inducible EFL1-expression vector treated (+) or not (−) with doxycycline. Arrows indicate increased 80S formation in complemented cells from individuals P1, P2, and P3. (M) Quantification of the 60S:80S ribosomal subunit ratios in cells transduced with inducible EFL1-expression vector treated (+) or not (−) with doxycycline (n = 3). (N) Inducible expression of wild-type EFL1 rescues global protein translation rates in EFL1-deficient fibroblast cell lines (n = 6). (O) Schematic of eIF6 release assay. Pre-60S subunits extracted from P3-derived fibroblasts were incubated with the indicated release factors and pelleted through a 15% sucrose cushion. Immunoblotting reveals the eIF6 distribution in the supernatant (“free”) and pellet (“bound”). (P) Release of eIF6 by SDS-associated EFL1 variants. P3-derived pre-60S subunits were incubated with guanosine triphosphate (GTP), SBDS, and the indicated EFL1 variants. EDTA was added as a positive control for eIF6 release. eIF6 and uL14 were visualized by immunoblotting. (−) indicates the negative control lacking EFL1. All data represent mean ± standard error. Statistical significance between samples was assessed by a 2-tailed Student t test. ns, not significant.
SDS-associated EFL1 mutations impair eIF6 release. (A) Total cell lysates from fibroblasts and (B) B-LCL from wild-type and 3 individuals (P1, P2, and P3) with SDS were immunoblotted to visualize the indicated proteins. (C) Defective ribosome assembly in EFL1 mutant fibroblasts. Polysome profiles from fibroblast extracts from 3 unrelated individuals with SDS compared with wild-type control. Quantification of the 60S:80S ribosomal subunit ratios is indicated as a bar chart (n ≥ 5) (D). EFL1 genotypes are provided in supplemental Table 1. (E) EFL1 is required for eIF6 recycling in human cells. Indicated proteins were visualized by immunoblotting in cytoplasmic or nuclear fractions from wild-type and EFL1 mutant fibroblasts. Histone H3, nuclear marker; HSP9, cytoplasmic marker. (F) Relative amount of eIF6 in the nucleus of EFL1 mutant cells compared with wild-type (n = 5). (G-H) EFL1 deficiency attenuates protein synthesis. OP-Puro incorporation in fibroblast cells lines from individuals P1, P2, and P3 relative to wild-type control cells quantified 1 hour after OP-Puro administration (n = 6). (I) Complementation of EFL1-deficient fibroblasts with wild-type EFL1. Lysates from EFL1 mutant fibroblasts transduced with a vector expressing GFP alone (+empty) or GFP + EFL1 (+EFL1) and from wild-type fibroblasts as a control. The indicated proteins were visualized by immunoblotting. (J) Wild-type EFL1 rescues global translation in patient fibroblast cell lines from P1 and P3 (n = 4). (K) Complementation of EFL1-deficient fibroblasts with inducible vector allows wild-type EFL1 expression after doxycycline (Dox) treatment. Lysates from EFL1 mutant fibroblasts transduced with an empty vector or EFL1-expressing vector with (+) or without (−) doxycycline. Vinculin is used as a loading control. (L) Comparison of polysome profiles from wild-type and EFL1-mutant fibroblasts transduced with empty vector or inducible EFL1-expression vector treated (+) or not (−) with doxycycline. Arrows indicate increased 80S formation in complemented cells from individuals P1, P2, and P3. (M) Quantification of the 60S:80S ribosomal subunit ratios in cells transduced with inducible EFL1-expression vector treated (+) or not (−) with doxycycline (n = 3). (N) Inducible expression of wild-type EFL1 rescues global protein translation rates in EFL1-deficient fibroblast cell lines (n = 6). (O) Schematic of eIF6 release assay. Pre-60S subunits extracted from P3-derived fibroblasts were incubated with the indicated release factors and pelleted through a 15% sucrose cushion. Immunoblotting reveals the eIF6 distribution in the supernatant (“free”) and pellet (“bound”). (P) Release of eIF6 by SDS-associated EFL1 variants. P3-derived pre-60S subunits were incubated with guanosine triphosphate (GTP), SBDS, and the indicated EFL1 variants. EDTA was added as a positive control for eIF6 release. eIF6 and uL14 were visualized by immunoblotting. (−) indicates the negative control lacking EFL1. All data represent mean ± standard error. Statistical significance between samples was assessed by a 2-tailed Student t test. ns, not significant.
Next, we tested if EFL1 mutations affect EFL1 function in ribosomal biogenesis. Consistent with a defect in ribosomal subunit joining, sucrose gradient centrifugation analysis of lysates from EFL1 mutant fibroblasts revealed an increased ratio of 60S:80S ribosomal subunits relative to control (Figure 3C-D). The severity of the ribosomal subunit joining defect correlated with the level of residual EFL1 protein expression in P1 and P3’s lysates (Figure 3A-B). Although the expression of mutant EFL1 protein in P2’s cells is similar to wild-type, the increased 60S:80S ratio in these cells indicates that there is functional impairment of the mutant EFL1 protein.
In wild-type cells, eIF6 binds to pre-60S ribosomal subunits in the nucleus and shuttles to the cytoplasm, where it is released during the terminal step in 60S subunit maturation. Thus, eIF6 is distributed between both the nuclear and cytoplasmic compartments. Compared with controls, the amount of eIF6 in the nucleus of EFL1 mutant fibroblasts was reduced by 70% to 80% (Figure 3E-F), consistent with a role for human EFL1 in promoting efficient nuclear recycling of eIF6.
As ribosomal subunit joining is impaired in EFL1 mutant fibroblasts derived from all 3 patients, we anticipated that global protein synthesis would be attenuated in these cells. Indeed, protein synthesis was reduced in fibroblasts from P1 (62%), P2 (50%), and P3 (51%) as assessed by in vivo incorporation of O-propargyl-puromycin (OP-Puro) (Figure 3G-H).21,32 To further link this translational defect with the EFL1 mutations, we cloned the wild-type EFL1 coding sequence into a lentiviral vector and transduced the EFL1 mutant fibroblasts from P1 and P3. The ectopic expression of wild-type EFL1 improved the translation rate in cells derived from P1 and P3 (Figure 3I-J). Likewise, the inducible expression of wild-type EFL1 in cells from P1, P2, and P3 (Figure 3K) improved both the ribosomal subunit joining defect as assessed by the 60S:80S ratio following sucrose gradient centrifugation (Figure 3L-M) and global translation (Figure 3N), as assessed by incorporation of the methionine analog azidohomoalanine.21 Lastly, we assessed the functional impact of the EFL1 missense mutations identified in P1, P2, and P3 by biochemically reconstituting eIF6 release ex vivo from human pre-60S ribosomal subunits. We added recombinant human wild-type or mutant EFL1 proteins together with SBDS and guanosine triphosphate to eIF6-loaded pre-60S subunits extracted from P3-derived human fibroblasts and monitored eIF6 removal by immunoblotting the “free” and “bound” fractions after pelleting the ribosomal subunits through a sucrose cushion (Figure 3O). Recombinant wild-type EFL1 efficiently triggered eIF6 release (Figure 3P). The p.C883G EFL1 mutant identified in individual P3 did not significantly impair eIF6 release compared with wild-type EFL1 (Figure 3P), suggesting that the combined defects in the polysome profile (Figure 3C-D), eIF6 subcellular localization (Figure 3E-F), and translation (Figure 3G-H) observed in the cells from P3 are a consequence of the strong reduction in EFL1 protein expression (Figure 3A-B). In sharp contrast, both the p.F505S and p.R970H EFL1 mutants identified in P1 and P2, respectively, yielded a markedly reduced amount of eIF6 in the free fraction compared with wild-type, indicating that their ability to remove eIF6 is impaired (Figure 3P).
We conclude from these results that the SDS-associated EFL1 mutations that we have identified are hypomorphic and cause the SDS phenotype by impairing (not completely abrogating) eIF6 release from late cytoplasmic 60S ribosomal subunits, thereby disrupting ribosomal subunit joining and attenuating global protein synthesis.
Efl1-mutant mouse recapitulates the SDS phenotype
To further assess the pathogenic effects of EFL1 mutations in vivo, we analyzed the Efl1-K983R mouse line (henceforth named Efl1K983R/K983R) generated by N-ethyl-N-nitrosourea–induced mutagenesis.33 Although the mouse mutation was not identified in any of the patients, Efl1 residue K983 (human K976) is highly conserved (Figure 4A) and lies within domain IV of EFL1, in close proximity to human residues R970 and C883 mutated in P2 and P3, respectively (Figure 1F). Most importantly, similar to the mutations identified in P1 and P3, the p.K983R substitution in the mouse causes a reduction in EFL1 protein expression as determined by immunoblotting (Figure 4B-C) while the total levels of eIF6 and SBDS were unaffected (data not shown). At the functional level, the murine p.K983R mutation also increased the 60S:80S ratio (Figure 4D-E), although the relative rate of protein synthesis was not significantly affected (Figure 4F). We conclude that the mouse p.K983R mutation is a valid model in which to study the broad phenotypic effects of EFL1 loss of function.
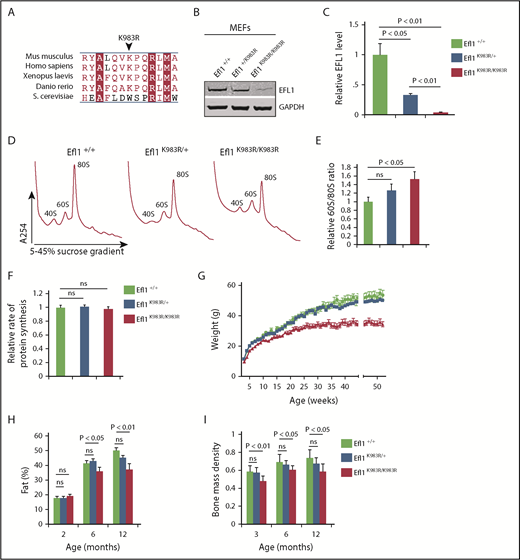
Efl1 K983R mutation leads to pleiotropic effects in mice. (A) Multiple sequence alignment of EFL1 proteins from representative species indicates that the murine EFL1 K983R mutation targets a highly conserved residue. Identical amino acids are indicated by a red box with white characters, similar amino acids are in red characters, and a blue frame represents similarity across groups. (B) Representative total cell lysates of mouse embryonic fibroblasts (MEFs) from wild-type (Efl1+/+), heterozygous Efl1+/K983R, and homozygous Efl1K983R /K983R mice were immunoblotted to visualize EFL1 protein. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is used as a loading control. (C) Quantifications show the EFL1/glyceraldehyde-3-phosphate dehydrogenase ratio. (D) Representative polysome profiles of MEF extracts from Efl1K983R /K983R (n = 4) and heterozygous Efl1+/K983R (n = 9) mice compared with wild-type control (Efl1+/+) (n = 9). (E) Quantification of the 60S:80S ribosomal subunit ratios is indicated as a bar chart (n ≥ 4). (F) Global protein translation rates in MEFs from Efl1K983R /K983R and heterozygous Efl1+/K983R mice compared with wild-type control (Efl1+/+) (n = 6). EFL1 K983R mutation reduces body weight from 3 weeks of age (G), affecting the percentage of fat mass (H) and bone mass density (I) from 3 months of age. For all tests, P values are indicated.
Efl1 K983R mutation leads to pleiotropic effects in mice. (A) Multiple sequence alignment of EFL1 proteins from representative species indicates that the murine EFL1 K983R mutation targets a highly conserved residue. Identical amino acids are indicated by a red box with white characters, similar amino acids are in red characters, and a blue frame represents similarity across groups. (B) Representative total cell lysates of mouse embryonic fibroblasts (MEFs) from wild-type (Efl1+/+), heterozygous Efl1+/K983R, and homozygous Efl1K983R /K983R mice were immunoblotted to visualize EFL1 protein. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is used as a loading control. (C) Quantifications show the EFL1/glyceraldehyde-3-phosphate dehydrogenase ratio. (D) Representative polysome profiles of MEF extracts from Efl1K983R /K983R (n = 4) and heterozygous Efl1+/K983R (n = 9) mice compared with wild-type control (Efl1+/+) (n = 9). (E) Quantification of the 60S:80S ribosomal subunit ratios is indicated as a bar chart (n ≥ 4). (F) Global protein translation rates in MEFs from Efl1K983R /K983R and heterozygous Efl1+/K983R mice compared with wild-type control (Efl1+/+) (n = 6). EFL1 K983R mutation reduces body weight from 3 weeks of age (G), affecting the percentage of fat mass (H) and bone mass density (I) from 3 months of age. For all tests, P values are indicated.
Efl1K983R/K983R mice were born at a normal Mendelian ratio and did not show any obvious pathological abnormalities after necropsy, including analysis of pancreas, liver, skeletal muscle, and brain. However, the mice exhibited a significant reduction in weight compared with wild-type or Efl1K983R/wt mice (Figure 4G), due to a progressive decrease in fat mass accumulation (Figure 4H). Although radiographs revealed no structural abnormalities of the skeleton in the Efl1K983R/K983R mice (data not shown), bone mass density was significantly reduced compared with wild-type littermates at different ages (Figure 4I). At the hematological level, compared with wild-type mice, the bone marrow cellularity (Figure 5A) as well as the frequency and absolute numbers of hematopoietic stem and progenitor cells (including megakaryocyte, granulocyte, erythroid progenitors; megakaryocyte progenitors; and pre–granulocyte-monocyte/granulocyte-monocyte progenitors34 ) were reduced in Efl1K983R/K983R mice (Figure 5B-C; supplemental Figure 4). Additionally, we noticed a significant reduction in hemoglobin (Figure 5D), platelet (Figure 5E), and total white cell counts (Figure 5F) in Efl1K983R/K983R mice.
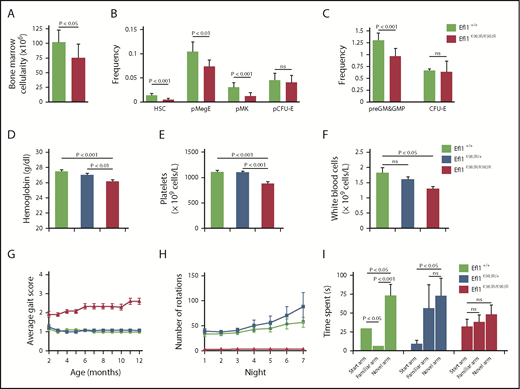
Impaired hematopoiesis, motor abnormalities, and cognitive deficits in Efl1K983R/K983Rmice. (A) Bone marrow cellularity in wild-type and homozygous Efl1K983R/K983R mice is indicated. (B-C) Frequencies of hematopoietic stem cells (HSCs); megakaryocyte, granulocyte, erythroid progenitors (pMegE); megakaryocyte progenitors (MkP); myeloid progenitors (pre–granulocyte-monocyte/granulocyte-monocyte progenitors); and erythroid progenitors (pCFU-E and CFU-E) in the bone marrow in the indicated genotypes are shown (n = 8 per genotype).34 EFL1 K983R mutation affects hematopoiesis. Hemoglobin levels (D), platelets (E), and total white blood cell counts (F). (G) Gait abnormalities in K983R mutant mice. Gait was analyzed using a qualitative scoring system at selected time points. Scores are explained in Materials and methods. (H) Motor deficits in EFL1 K983R mutant mice. Free wheel-running activity (over 7 nights, 3 months of age) for the indicated genotypes. (I) Cognitive deficits in K983R mutant mice. Y maze habituation testing of the indicated genotypes at 3 months of age. For all tests, P values are indicated.
Impaired hematopoiesis, motor abnormalities, and cognitive deficits in Efl1K983R/K983Rmice. (A) Bone marrow cellularity in wild-type and homozygous Efl1K983R/K983R mice is indicated. (B-C) Frequencies of hematopoietic stem cells (HSCs); megakaryocyte, granulocyte, erythroid progenitors (pMegE); megakaryocyte progenitors (MkP); myeloid progenitors (pre–granulocyte-monocyte/granulocyte-monocyte progenitors); and erythroid progenitors (pCFU-E and CFU-E) in the bone marrow in the indicated genotypes are shown (n = 8 per genotype).34 EFL1 K983R mutation affects hematopoiesis. Hemoglobin levels (D), platelets (E), and total white blood cell counts (F). (G) Gait abnormalities in K983R mutant mice. Gait was analyzed using a qualitative scoring system at selected time points. Scores are explained in Materials and methods. (H) Motor deficits in EFL1 K983R mutant mice. Free wheel-running activity (over 7 nights, 3 months of age) for the indicated genotypes. (I) Cognitive deficits in K983R mutant mice. Y maze habituation testing of the indicated genotypes at 3 months of age. For all tests, P values are indicated.
Some of the EFL1-deficient individuals suffered from motor abnormalities and cognitive deficits also present in the Efl1K983R/K983R mutant mice. Indeed, we identified gait abnormalities (Figure 5G) and motor dysfunction (Figure 5H) when compared with littermate controls. Moreover, working memory was also disrupted in Efl1K983R/K983R mice, as tested via Y maze habituation (Figure 5I).
We conclude from these observations that Efl1K983R/K983R mutant mice recapitulate key aspects of the SDS phenotype, strongly supporting a causal role for EFL1 dysfunction in SDS.
Discussion
In this study, we provide compelling evidence that recessive EFL1 mutations cause SDS by impairing release of eIF6 from nascent cytoplasmic 60S ribosomal subunits, thereby disrupting ribosomal subunit joining and attenuating global protein synthesis. Taken together with previous work,22-24 our human genetic and biochemical data indicate that the release of eIF6 by the concerted action of EFL1 and its allosteric activator, SBDS, is a fundamental conserved mechanism that licenses the entry of functionally competent large ribosomal subunits into active translation. These data do not support a critical role for RACK1 and PKCβII in the eviction of eIF6 in mammalian cells through the phosphorylation of eIF6 residue S235 as previously proposed.11 Indeed, the position of RACK1 on the head of the 40S subunit,35 the viability and normal growth of PKCβ null mutant mice,36 and the observation that the phylogenetically conserved eIF6 residues 1 to 224 are sufficient for eIF6 recycling in vivo4 are inconsistent with such a model. Our work provides the most compelling evidence to date that SDS is a ribosomopathy that is caused by mutations in multiple components (SBDS, EFL1, and DNAJC21) of a coherent pathway involved in cytoplasmic maturation of the 60S ribosomal subunit maturation.
Through our analysis, we identified an EFL1 pseudogene (EFL1P1; chromosome 15: 84 080,168-84 126,604) that is located 1.8 Mb distal to the EFL1 gene and is directionally inverted (supplemental Figure 5A). This is striking, because in 89% of cases, SBDS mutations arise from a gene conversion event due to recombination between SBDS and its pseudogene (SBDSP), located 5.8 Mb distally in an inverted orientation.37,38 Thus, the c.1514T>C (p.F505S) mutation identified in P1 might arise from gene conversion due to recombination between EFL1 and the EFL1P1 pseudogene containing this sequence change (supplemental Figure 5B-C). The genetic analysis of EFL1 is further challenged by the existence of a second EFL1 pseudogene (EFL1P2) located on a distinct chromosome (chromosome 4: 65 142,703-65 145,763), as well as several segmental duplications located on chromosomes 15 and Y (supplemental Table 2). These numerous genomic sequences that are highly homologous to the EFL1 gene may hamper the identification of pathological variants in EFL1. Furthermore, an uncharacterized genetic event in P3 has resulted in a profound reduction in EFL1 expression from the maternal allele (Figure 1A, zoom; supplemental Figure 2).39,40 This observation suggests that analysis of the EFL1 coding sequence in SDS patients who lack SBDS mutations may be insufficient to detect the genetic events leading to defective EFL1 expression. We therefore recommend that EFL1 mutation status and EFL1 protein expression should be routinely assessed in individuals with SDS lacking mutations in SBDS and DNAJC21.
During the course of our study, Stepensky et al41 and Tan et al42 investigated a total of 6 individuals carrying biallelic variants in the EFL1-encoding gene. However, the pathological significance of these mutations remains unclear, as introduction of the corresponding disease-related variants in yeast had no significant effect on cell fitness41 and the functional impact of the reported EFL1 missense variants on human ribosome assembly and translation was not assessed.41,42
Autosomal dominant mutations in SRP54, a key member of the cotranslational protein-targeting pathway, results in neutropenia associated with SDS-like features such as pancreatic insufficiency and short stature.43,44 However, myeloid maturation arrest at the promyelocyte stage and the presence of prominent cytoplasmic vacuoles in the myeloid precursors are not typical of classical SDS. Thus, the mechanism underlying the neutropenia may differ in patients with SRP54 mutations compared with that in typical SDS patients.
Structural, biochemical, and genetic studies4,22-24,45 support a cofactor-dependent conformational-switching mechanism for release of the antiassociation factor eIF6 from the intersubunit face of the 60S subunit. This model proposes that SBDS drives the conformational equilibrium of EFL1 toward a high-affinity state that effectively competes with eIF6 for an overlapping binding site on the sarcin-ricin loop of the 60S subunit to promote eIF6 displacement. Supporting this hypothesis, cells derived from individuals carrying EFL1 mutations exhibit defects in eIF6 release and recycling, ribosomal subunit joining, and protein translation that were corrected by expressing the wild-type EFL1 protein (Figure 3). The EFL1 p.C883G (P3) and murine p.K983R (human K976) mutations appear to simply reduce EFL1 expression. However, the EFL1 p.R970H mutation (P2) that is expressed at wild-type levels is functionally defective. Interestingly, like residues C883 and K970, K976 maps to the interface between EFL1 domain IV (essential for Efl1 function in vivo in yeast29 ) and the C-terminal domain of SBDS (Figure 1F), where it may potentially disrupt the allosteric activation of EFL1 by SBDS. Furthermore, the EFL1 variants p.M882K and p.R1095Q reported by Stepensky et al41 map to this interface. The EFL1 p.F505S mutation (P1) is also functionally impaired, likely disrupting a key dynamic interface between EFL1 domains II and III that mediates EFL1 conformational change on the ribosome.4 However, further investigations are required to precisely delineate the underlying mechanisms.
We reinforce these data by characterizing a novel mouse model carrying a point mutation within the Efl1 gene. Like the human EFL1 mutations identified here, the EFL1 p.K983R mouse mutation leads to reduced EFL1 protein expression, resulting in defects in ribosomal maturation. The mutation models key aspects of the human disease, including hematological abnormalities and growth deficits, as well as the defects in motor and cognitive function that are also present in some of the patients identified here, strongly supporting the conclusion that the SDS phenotype is caused by loss of EFL1 function.
How do we explain the striking tissue proclivity in SDS? Our data demonstrate that similar to SBDS deficiency,24,46,47 impaired EFL1 function markedly attenuates global protein synthesis by impairing ribosomal subunit joining due to the reduced availability of free cytoplasmic 60S ribosomal subunits. Therefore, the SDS phenotype likely reflects at least in part the impaired capacity of specific tissues to upregulate global protein translation at key time points during development (and indeed postnatally) due to inefficient recruitment of nascent 60S subunits into the actively translating pool. Impaired translation reinitiation of complex messenger RNAs carrying upstream open reading frames may also contribute to the SDS phenotype.48
Tissue-specific sensitivities to cellular stress responses evoked by altered ribosome homeostasis may not only contribute to the SDS disease phenotype49 but may also potentially drive cancer progression by promoting the selection of compensatory mutations.50 Thus, the somatic acquisition of biallelic TP53 variants in SBDS-deficient hematopoietic stem and progenitor cells is associated with clonal progression and transformation to poor-prognosis MDS.5,50 Furthermore, recurrent acquired interstitial deletions of chromosome 20 in SDS bone marrow cells encompass the EIF6 gene,51,52 providing a potential mechanism that allows SDS cells to bypass the defect in ribosomal subunit joining by reducing the cellular dose of eIF6. Whether eIF6 deletion indeed modulates the risk of acquiring somatic TP53 variants and progression to MDS and acute myeloid leukemia is an interesting question that will require careful clinical follow up of all genetic subgroups of SDS. Our data support the hypothesis that the development of small molecules that mimic the effect of TIF6 suppressor mutations in yeast24 may have therapeutic potential in SDS.
For original data, please contact either Patrick Revy (patrick.revy@inserm.fr) or Alan. J. Warren (ajw1000@cam.ac.uk).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the patients and their families for their contribution in this study. P.R. is grateful to Alain Fischer for discussions, advice, and support. The authors acknowledge the excellent technical assistance of Alicia Fernandes, who generated B-LCLs (Centre de Ressource Biologique, Imagine Institute, Paris, France). The authors thank F. Fouyssac, N. Aladjidi, V. Barlogis, and F. Monpoux for patient care.
P.J. was funded by the Swedish Research Council (2014-06807). This work was supported by institutional grants from INSERM, Ligue Nationale contre le Cancer (Equipe Labellisée La Ligue), and GIS-Institut des maladies rares and State funding from the Agence Nationale de la Recherche under “Investissements d’avenir” program (ANR-10-IAHU-01). P.R. is a scientist from Centre National de la Recherche Scientifique. The work was also supported by a Specialist Programme from Bloodwise (12048 [A.J.W.]), the UK Medical Research Council (MC_U105161083 [A.J.W.]; MC_UP_A390_1106 and 1330931 [A.A.-A.]), a Wellcome Trust strategic award to the Cambridge Institute for Medical Research (100140), a core support grant from the Wellcome Trust and Medical Research Council to the Wellcome Trust-Medical Research Council Cambridge Stem Cell Institute, Ted’s Gang and The Connor Wright Shwachman Diamond Project (A.J.W.), and the Cambridge National Institute for Health Research Biomedical Research Centre.
Authorship
Contribution: C.K. and P.R. initiated the project and performed whole-exome sequencing analysis; P.R. and A.J.W. supervised and coordinated the project; C.K., M.P., O.F., H.D.l.P., B.B., E.L., J.D., D.R., and C.B.-C. identified affected patients or assisted with related clinical, genetic, and laboratory studies. Sanger sequencing and cloning were performed by L.K., B.B., C.K., and P.R.; EFL1 constructs were generated by P.R.; L.K. performed transduction of the wild-type EFL1 expression vectors.; A.F. and S.T. devised and performed ex vivo eIF6 release assays. A.A.-A., A.H., P.J., and A.J.W. generated and performed phenotypic analyses of the Efl1 mutant mice; S.T. performed functional characterization of patient cell lines; S.F. initiated functional characterization of patient cell lines; P.N. performed bioinformatics analysis; C.B.-F. performed next-generation sequencing experiments; and P.R. and A.J.W. prepared figures and wrote the manuscript, supported by S.T., C.K., J.D., C.B.-C., E.L., J.-P.d.V., P.J., A.F., and A.A.-A.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Patrick Revy, Imagine Institute for Genetic Diseases, 24 Blvd du Montparnasse, Paris 75015, France; e-mail: patrick.revy@inserm.fr; and Alan J. Warren, Cambridge Institute for Medical Research, Keith Peters Building, Hills Rd, Cambridge CB2 0XY, United Kingdom; e-mail: ajw1000@cam.ac.uk.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal