


Abstract
Waldenström macroglobulinemia (WM) is an uncommon lymphoma characterized by the infiltration of the bone marrow by clonal lymphoplasmacytic cells that produce monoclonal immunoglobulin M (IgM). The disease may have an asymptomatic phase, or patients may present with symptoms and complications resulting from marrow or other tissue infiltration, or from physicochemical or immunological properties of the monoclonal IgM. Diagnosis of WM has been clearly defined, and genetic testing for somatic mutation of MYD88L265P is a useful tool for differential diagnosis from other conditions. Specific criteria that define symptomatic disease that needs treatment offer clinical guidance. The treatment of WM has evolved rapidly, with treatment options that include anti-CD20 monoclonal antibody-based combinations and BTK inhibitors. The choice of therapy is based on the need for rapid disease control, presence of specific disease complications, and patient’s age. With the use of BTK inhibitors, the use of continuous therapy has been introduced as another option over fixed-duration chemoimmunotherapy. In this review, we focus on different clinical scenarios and discuss treatment options, based on the available data.
Introduction
Waldenström macroglobulinemia (WM) is an uncommon lymphoma (∼1%-2% of hematological malignancies) with unique features, characterized by the accumulation of lymphoplasmacytic cells that produce monoclonal immunoglobulin M (IgM). Symptoms and complications are related to tumor burden or quantity or to the physicochemical or immunological properties of monoclonal IgM (Table 1), but the disease also may have a long asymptomatic course. WM is mostly a disease of the elderly1,2 and has higher prevalence among whites1 and a familial predisposition.3,4
Clinical and laboratory findings of symptomatic WM
| Characteristics of patients with symptomatic WM | N = 595 |
|---|---|
| Age, median/range, y | 69 (24-92) |
| Male/female, % | 60/40 |
| BM involvement, median | 52% |
| MYD88 L265P* (N = 84) | 77% |
| IgM, median | 3480 mg/dL |
| IgG, median | 790 mg/dL |
| IgA, median | 85 mg/dL |
| Hemoglobin, median | 10.1 g/dL |
| Hemoglobin <10 g/dL | 46% |
| Platelets, ×109/L, median | 215 |
| Platelets, <100 × 109/L | 12% |
| WBC, ×109/L, median | 6.6 |
| b2-microglobulin, median | 3.36 mg/dL |
| Serum albumin | 3.6 g/dL |
| Serum albumin <3.5 g/dL | 40% |
| LDH, U/L (ULN < 225 IU/L), median | 180 |
| LDH > ULN | 20% |
| Cryoglobulins present | 5.5% |
| Cold agglutinins present | 4% |
| Lymphadenopathy | 36% |
| Splenomegaly | 29% |
| Clinical presentation at the time of symptomatic disease: main indication for therapy (in many patients more than 1 reason was present) | |
| Anemia/cytopenias | 42% |
| B-symptoms | 25% |
| Hyperviscosity | 17% |
| Neuropathy | 12% |
| Amyloidosis | 1.5% |
| Symptomatic cryoglobulinemia | 1.3% |
| Symptomatic cold agglutinin disease | 0.6% |
| Other | 0.6% |
| Characteristics of patients with symptomatic WM | N = 595 |
|---|---|
| Age, median/range, y | 69 (24-92) |
| Male/female, % | 60/40 |
| BM involvement, median | 52% |
| MYD88 L265P* (N = 84) | 77% |
| IgM, median | 3480 mg/dL |
| IgG, median | 790 mg/dL |
| IgA, median | 85 mg/dL |
| Hemoglobin, median | 10.1 g/dL |
| Hemoglobin <10 g/dL | 46% |
| Platelets, ×109/L, median | 215 |
| Platelets, <100 × 109/L | 12% |
| WBC, ×109/L, median | 6.6 |
| b2-microglobulin, median | 3.36 mg/dL |
| Serum albumin | 3.6 g/dL |
| Serum albumin <3.5 g/dL | 40% |
| LDH, U/L (ULN < 225 IU/L), median | 180 |
| LDH > ULN | 20% |
| Cryoglobulins present | 5.5% |
| Cold agglutinins present | 4% |
| Lymphadenopathy | 36% |
| Splenomegaly | 29% |
| Clinical presentation at the time of symptomatic disease: main indication for therapy (in many patients more than 1 reason was present) | |
| Anemia/cytopenias | 42% |
| B-symptoms | 25% |
| Hyperviscosity | 17% |
| Neuropathy | 12% |
| Amyloidosis | 1.5% |
| Symptomatic cryoglobulinemia | 1.3% |
| Symptomatic cold agglutinin disease | 0.6% |
| Other | 0.6% |
Data are from the database of the Greek Myeloma Study Group.
Case 1
A 51-year-old otherwise healthy man was found to have high erythrocyte sedimentation rate during routine testing; a serum protein electrophoresis (SPEP) revealed a monoclonal IgM(κ) of 4.1 g/dL.
Diagnosis
The diagnosis of WM requires a bone marrow (BM) biopsy showing infiltration by clonal lymphoplasmacytic cells/lymphoplasmacytic lymphoma (LPL) and the presence of any amount of monoclonal IgM,5,6 detected by immunofixation electrophoresis. There is no threshold for BM clonal cell infiltration, but individuals with less than 10% clonal cells have an indolent course7 compared with those with at least 10% infiltration.8 Two types of clonal cells (B cells and plasma cells with varying degree of differentiation5,9 ) are usually found. A LPL without monoclonal IgM present or secreting monoclonal non-IgM is not WM, although the biology may not differ significantly. A monoclonal IgM without LPL histopathology in the BM is not WM, but either a monoclonal gammopathy of undetermined significance (MGUS) or a nodal lymphoma without BM infiltration. Patients not fulfilling WM criteria may still need treatment of IgM-related complications. Table 2 shows the typical WM morphology and immunophenotype5,6,9 and differential diagnosis. In 80% to 95% of patients, the lymphoplasmacytes harbor a somatic mutation in the myeloid differentiation primary response gene (MYD88L265P)10 ; however, frequency varies according to detection method and DNA source (whole BM, CD19-selected cells, paraffin-embedded tissue, peripheral blood, etc).11,12 Detection in peripheral blood using cell-free DNA is feasible, although with less sensitivity than in the BM.13 MYD88L265P detection is helpful to differentiate WM from morphologically similar lymphomas or IgM myeloma, but MYD88L265P alone is not diagnostic of WM. Absence of MYD88L265P does not exclude WM: 5% to 10% of patients with WM do not have MYD88L265P (they have other MYD88 mutations14 or have wild-type MYD88). MYD88L265P also is found in 30% to 80% of IgM MGUS cases10,13,15 (depending on method’s sensitivity) and in other lymphomas, but at significantly lower rates. In 20% to 40% of patients, lymphoplasmacytes have somatic activating mutations in the C-terminal domain of the C-X-C chemokine receptor type 4 (CXCR4) gene,10,16 which are similar to germline mutations observed in WHIM syndrome (CXCR4WHIM). These heterogeneous mutations may be either truncating or frameshift, with potentially different clinical effect,17 but are not helpful for WM diagnosis.
Differential diagnosis of WM from other diseases that may share a similar phenotype
| BM biopsy | Cytogenetics | MYD88L265P | Immunophenotype (by IHC or flow cytometry in BM) | Clinical presentation | |
|---|---|---|---|---|---|
| WM | Morphology: lymphoplasmacytes or cells with lymphoplasmacytic differentiation, together with a small population of clonal plasma cells ≥10% LPL* | Del6q (30%-50%) | 70%-90% | B-cell population: CD20+, sIgM+, CD22 + (weak), CD79+, CD25+, CD27+, FMC7+, BCL-2+, CD52+, CD5+/−, CD10+/−, CD23+/−, CD103−; plasma cell population: CD138+ CD38++, CD19+, CD45+, CD56− | Hyperviscosity, lymphadenopathy, splenomegaly, neuropathy |
| IgM MGUS | <10% LPL in the BM and <3 g/dL IgM* | ? | 30%-60% | Usually few cells found | No symptoms or only IgM-related |
| Myeloma | Plasma cells | t(11;14) or other IgH translocations | 0 | CD138+, CD38+, CD19− | Lytic bone disease Cyclin D1 staining positive in 75% [usually associated with t(11;14)] |
| SMZL | Intrasinusoidal infiltration by CD20+ cells | Del7q (19%), +3q(19%), +5q(10%) | 10% | CD19+, CD20+, CD22+, CD79a+, CD79b+, FMC7+ IgM+ CD5− (weakly + in 10%-25%), CD10−, CD43−, BCL6−, cyclin D1−CD103−, but occasionally + CD11c−/+ CD25−/+CD11c+ | Splenomegaly more common; circulating cells of characteristic morphology may be found |
| Follicular lymphoma | Small cleaved lymphocytes, paratrabecular localization in the BM | Translocations involving BCL-2 (70-90%) | 0 | CD5−, CD10+/−, CD11c−/+, CD103−, CD25−, CD138−, CD38+, CD45+, bcl2+, bcl6+ | Lymphadenopathy predominates |
| Mantle cell lymphoma | Monotypic, medium-small-sized lymphocytes with abnormal nucleus | t(11;14)(q13;q32) | 0 | CD5+, CD10−, CD23−, CD25−, CD45+, CD103−, CD138− | Lymphadenopathy and extranodal involvement common |
| BM biopsy | Cytogenetics | MYD88L265P | Immunophenotype (by IHC or flow cytometry in BM) | Clinical presentation | |
|---|---|---|---|---|---|
| WM | Morphology: lymphoplasmacytes or cells with lymphoplasmacytic differentiation, together with a small population of clonal plasma cells ≥10% LPL* | Del6q (30%-50%) | 70%-90% | B-cell population: CD20+, sIgM+, CD22 + (weak), CD79+, CD25+, CD27+, FMC7+, BCL-2+, CD52+, CD5+/−, CD10+/−, CD23+/−, CD103−; plasma cell population: CD138+ CD38++, CD19+, CD45+, CD56− | Hyperviscosity, lymphadenopathy, splenomegaly, neuropathy |
| IgM MGUS | <10% LPL in the BM and <3 g/dL IgM* | ? | 30%-60% | Usually few cells found | No symptoms or only IgM-related |
| Myeloma | Plasma cells | t(11;14) or other IgH translocations | 0 | CD138+, CD38+, CD19− | Lytic bone disease Cyclin D1 staining positive in 75% [usually associated with t(11;14)] |
| SMZL | Intrasinusoidal infiltration by CD20+ cells | Del7q (19%), +3q(19%), +5q(10%) | 10% | CD19+, CD20+, CD22+, CD79a+, CD79b+, FMC7+ IgM+ CD5− (weakly + in 10%-25%), CD10−, CD43−, BCL6−, cyclin D1−CD103−, but occasionally + CD11c−/+ CD25−/+CD11c+ | Splenomegaly more common; circulating cells of characteristic morphology may be found |
| Follicular lymphoma | Small cleaved lymphocytes, paratrabecular localization in the BM | Translocations involving BCL-2 (70-90%) | 0 | CD5−, CD10+/−, CD11c−/+, CD103−, CD25−, CD138−, CD38+, CD45+, bcl2+, bcl6+ | Lymphadenopathy predominates |
| Mantle cell lymphoma | Monotypic, medium-small-sized lymphocytes with abnormal nucleus | t(11;14)(q13;q32) | 0 | CD5+, CD10−, CD23−, CD25−, CD45+, CD103−, CD138− | Lymphadenopathy and extranodal involvement common |
SMZL, splenic marginal zone lymphoma.
This classification follows the proposal of Kyle et al.8 Per Consensus criteria5 and World Health Organization definitions,6 there is no threshold for the BM infiltration by clonal cells to define WM.5 Individuals with less than 10% clonal cells have an indolent course7 similar to that of MGUS compared with those with at least 10% LPL infiltration who have a higher risk for progression to symptomatic WM.8 However, patients not fulfilling WM criteria may still need treatment of the management of IgM-related complications.
Initial work-up
Table 3 shows the tests that could be helpful in patients with WM. Anemia or anemia-related fatigue should be evaluated as to whether they are a result of WM or other reasons (eg, iron deficiency18 ). Lactate dehydrogenase (LDH), serum albumin, and β-2 microglobulin are of prognostic significance. Evaluation of monoclonal IgM and other immunoglobulins are essential; uninvolved immunoglobulins are often suppressed.19 Determination of IgM is more accurate with densitometry than total serum IgM by nephelometry20 ; the same method should be used for comparisons and evaluation of response. Serum-free light chains are commonly altered and may be more useful to follow patients that developed light chain (AL) amyloidosis. Renal dysfunction may be present, and several renal pathologies have been described.21,22 Hyperviscosity syndrome related to high IgM levels is a hallmark of symptomatic WM, but there is no linear association of IgM levels with serum viscosity and hyperviscosity symptoms (headaches, blurred vision, confusion, epistaxis, gingival hemorrhages). A funduscopic examination is more reliable for detection of clinically significant hyperviscosity.23
Initial clinical and laboratory evaluation of WM
| Comments | |
|---|---|
| Clinical evaluation | |
| History and physical examination | Headache, blurred vision: consider HVS |
| Familial history for WM and other B-cell lymphoproliferative disorders | Skin rash: cryoglobulinemia (palpable purpura), Schnitzler syndrome (urticaria, bone pain, fever) |
| Review of systems for the presence of B symptoms, organomegaly, hyperviscosity symptoms, neuropathy, Raynaud’s disease, rash, peripheral edema, skin abnormalities, dyspnea | Symptoms of peripheral neuropathy: consider neurologist consultation |
| Funduscopic examination by an experienced ophthalmologist if IgM is high (ie, >3000 mg/dL) or hyperviscosity is suspected; photographic documentation may be useful for appreciation of future changes | Dyspnea, edema: consider amyloidosis |
| Laboratory evaluation | |
| Complete blood count | Additional tests may be required for evaluation of anemia, especially if it is the only indication of symptomatic disease (consider other causes such as iron deficiency or other) |
| Complete metabolic panel (including LDH, serum albumin) | Hemolysis should be considered if increased bilirubin and LDH |
| Serum Ig levels (IgA, IgG, IgM) | Urine evaluation at baseline is advised; if renal dysfunction or proteinuria is present, consider additional tests (cryoglobulins, FLCs etc) |
| Serum and urine electrophoresis with immunofixation | |
| Serum B2M level | |
| Viral serology (hepatitis B and C and HIV) | |
| Histology and molecular tests | |
| BM aspiration and biopsy | MYD88 testing not standardized; |
| IHC (required for diagnosis) | BM is preferable for testing MYD88, but other methods may also be useful |
| Flow cytometry (optional; consider if IHC not available) | The laboratory should report the sensitivity of MYD88 detection |
| Testing for MYD88L265P | Testing for CXCR4WHIM not helpful for diagnosis; not standardized, should be considered optional and not used for clinical decisions |
| There are limited data on the prognostic effect of cytogenetics by interphase fluorescence in situ hybridization or karyotype, although in some cases it may be helpful in differential diagnosis in addition to other findings | |
| Optional tests, if clinically indicated | |
| In case of Raynaud’s, renal dysfunction, hematuria, skin rash, hyperviscosity consider evaluation for cryoglobulins | Cryoglobulins may require special communication with the laboratory |
| Hemolysis, hyperviscosity: consider cold agglutinin titer | In the presence of cryoglobulins, the assessment of IgM and response may be challenging |
| Serum viscosity (not always correlated with symptoms) | |
| Bleeding diathesis with prolonged aPTT and PT: screening for acquired von Willebrand disease | |
| Suspicion of amyloidosis: 24-hour urine protein quantification, Serum FLCs, NTproBNP, Cardiac troponins | |
| Symptoms of peripheral neuropathy are reported: nerve conduction studies, myelin-associated globulin antibodies, anti-ganglioside M1, other antibodies (consultation with neurologist strongly advised) | |
| Central nervous system symptoms: consider Bing-Neel syndrome, brain/spine magnetic resonance imaging, cerebrospinal fluid testing (also for MYD88L265P) | |
| Renal dysfunction: consider renal biopsy if indicated. Several renal pathologies have been described such as amyloidosis, cryoglobulinemic glomerulonephritis, immunoglobulin deposition disease, cast nephropathy, etc21,22 |
| Comments | |
|---|---|
| Clinical evaluation | |
| History and physical examination | Headache, blurred vision: consider HVS |
| Familial history for WM and other B-cell lymphoproliferative disorders | Skin rash: cryoglobulinemia (palpable purpura), Schnitzler syndrome (urticaria, bone pain, fever) |
| Review of systems for the presence of B symptoms, organomegaly, hyperviscosity symptoms, neuropathy, Raynaud’s disease, rash, peripheral edema, skin abnormalities, dyspnea | Symptoms of peripheral neuropathy: consider neurologist consultation |
| Funduscopic examination by an experienced ophthalmologist if IgM is high (ie, >3000 mg/dL) or hyperviscosity is suspected; photographic documentation may be useful for appreciation of future changes | Dyspnea, edema: consider amyloidosis |
| Laboratory evaluation | |
| Complete blood count | Additional tests may be required for evaluation of anemia, especially if it is the only indication of symptomatic disease (consider other causes such as iron deficiency or other) |
| Complete metabolic panel (including LDH, serum albumin) | Hemolysis should be considered if increased bilirubin and LDH |
| Serum Ig levels (IgA, IgG, IgM) | Urine evaluation at baseline is advised; if renal dysfunction or proteinuria is present, consider additional tests (cryoglobulins, FLCs etc) |
| Serum and urine electrophoresis with immunofixation | |
| Serum B2M level | |
| Viral serology (hepatitis B and C and HIV) | |
| Histology and molecular tests | |
| BM aspiration and biopsy | MYD88 testing not standardized; |
| IHC (required for diagnosis) | BM is preferable for testing MYD88, but other methods may also be useful |
| Flow cytometry (optional; consider if IHC not available) | The laboratory should report the sensitivity of MYD88 detection |
| Testing for MYD88L265P | Testing for CXCR4WHIM not helpful for diagnosis; not standardized, should be considered optional and not used for clinical decisions |
| There are limited data on the prognostic effect of cytogenetics by interphase fluorescence in situ hybridization or karyotype, although in some cases it may be helpful in differential diagnosis in addition to other findings | |
| Optional tests, if clinically indicated | |
| In case of Raynaud’s, renal dysfunction, hematuria, skin rash, hyperviscosity consider evaluation for cryoglobulins | Cryoglobulins may require special communication with the laboratory |
| Hemolysis, hyperviscosity: consider cold agglutinin titer | In the presence of cryoglobulins, the assessment of IgM and response may be challenging |
| Serum viscosity (not always correlated with symptoms) | |
| Bleeding diathesis with prolonged aPTT and PT: screening for acquired von Willebrand disease | |
| Suspicion of amyloidosis: 24-hour urine protein quantification, Serum FLCs, NTproBNP, Cardiac troponins | |
| Symptoms of peripheral neuropathy are reported: nerve conduction studies, myelin-associated globulin antibodies, anti-ganglioside M1, other antibodies (consultation with neurologist strongly advised) | |
| Central nervous system symptoms: consider Bing-Neel syndrome, brain/spine magnetic resonance imaging, cerebrospinal fluid testing (also for MYD88L265P) | |
| Renal dysfunction: consider renal biopsy if indicated. Several renal pathologies have been described such as amyloidosis, cryoglobulinemic glomerulonephritis, immunoglobulin deposition disease, cast nephropathy, etc21,22 |
FLC, free light chains; HVS, hyperviscosity syndrome.
Peripheral neuropathy is common, and often the only indication to start treatment in otherwise asymptomatic patients. Neuropathy is usually sensory, symmetrical, ascending, starting from the feet, demyelinating,24 and typically slowly progressing. Rapidly progressing neuropathy should alert for alternate causes. High serum titters of myelin-associated globulin antibodies are found in ∼50% of these patients.25 Less often, anti-ganglioside M1 antibodies may be found, but in such cases, motor neuropathy predominates. Nerve conduction studies may show axonal degeneration in patients with longstanding sensorimotor neuropathy or amyloidosis; small fiber neuropathy may also be seen. There may be other unrelated causes for neuropathy, especially in older patients, and consultation with a neurologist is advised. Amyloidosis may complicate WM, affecting kidneys, heart, liver, and nerves26 ; AL is the most common type in WM,26 but others (such as AA amyloidosis) have been described. In the case of isolated cardiac involvement in an elderly male patients with WM, evaluation for ATTRwt should also be performed.27 Computed tomography (CT) or magnetic resonance imaging are useful for evaluation of organomegaly and lymphadenopathy. Positron emission tomography does not seem to offer additional information,28 but if transformation to an aggressive lymphoma or another malignancy is suspected, positron emission tomography/CT may be used to biopsy the most fluorodeoxyglucose–avid lesion.
Case 1
Patient’s hemoglobin is 14.2 g/dL, and he has no B symptoms, splenomegaly, or lymphadenopathy. A BM biopsy showed 50% infiltration by lymphoplasmacytes; molecular testing revealed MYD88L265P.
Indications for therapy
About 19% to 28% of patients have asymptomatic WM8,29 and can remain asymptomatic for several years; median time to symptom development may exceed 5 to 10 years.8,30,31 Table 4 depicts indications to start therapy31,32 ; however, clinical judgement is required. The level of monoclonal IgM alone is not an indication to start treatment31,32 ; however, among those with high IgM levels (>6000 mg/dL), data are conflicting.33,34 Our approach is to follow such patients closely. Recently, a score based on BM infiltration percentage, IgM levels, b2-microglobulin, and albumin was developed that identified 3 groups of asymptomatic WM with a median time to development of symptomatic disease of 1.8, 4.8, and 9.3 years.7 There are no data to support early initiation of therapy over a watch-and-wait strategy, even in patients at high risk for progression; such patients should be managed in clinical trials. The patient in case 1 was asymptomatic at initial evaluation and, according to the score above, at intermediate risk. A close follow-up and clinical evaluation is important, and he was followed clinically at 3-month intervals, at least for the first couple of years, to evaluate the pace of the disease.
Indications to start therapy in a patient with a diagnosis of WM
| Clinical indications for initiation of therapy |
|---|
| Recurrent fever, night sweats, weight loss, fatigue |
| Hyperviscosity |
| Lymphadenopathy: either symptomatic or bulky (≥5 cm in maximum diameter) |
| Symptomatic hepatomegaly and/or splenomegaly |
| Symptomatic organomegaly and/or organ or tissue infiltration |
| Peripheral neuropathy because of WM |
| Clinical indications for initiation of therapy |
|---|
| Recurrent fever, night sweats, weight loss, fatigue |
| Hyperviscosity |
| Lymphadenopathy: either symptomatic or bulky (≥5 cm in maximum diameter) |
| Symptomatic hepatomegaly and/or splenomegaly |
| Symptomatic organomegaly and/or organ or tissue infiltration |
| Peripheral neuropathy because of WM |
| Laboratory indications for initiation of therapy |
|---|
| Symptomatic cryoglobulinemia |
| Symptomatic cold agglutinin anemia |
| Autoimmune hemolytic anemia and/or thrombocytopenia |
| Nephropathy that is related to WM |
| Amyloidosis that is related to WM |
| Hemoglobin ≤10 g/dL |
| Platelet count <100 × 109/L |
| Laboratory indications for initiation of therapy |
|---|
| Symptomatic cryoglobulinemia |
| Symptomatic cold agglutinin anemia |
| Autoimmune hemolytic anemia and/or thrombocytopenia |
| Nephropathy that is related to WM |
| Amyloidosis that is related to WM |
| Hemoglobin ≤10 g/dL |
| Platelet count <100 × 109/L |
Risk assessment in symptomatic WM
A prognostic system International Prognostic Scoring System for WM (IPSSWM), based on age, b2-microglobulin, hemoglobin, platelet counts, and IgM level, stratifies patients into 3 risk groups.35 Recently, a revised score (rIPSSWM) was developed36 on the basis of age, b2-microglobulin, serum LDH, and albumin, and identifies 5 prognostic groups. In IPSSWM and rIPSSWM, age is a critical prognostic determinant, and both are based on biochemical parameters and not molecular or genetic characteristics. There are limited data on the prognostic effect of cytogenetics by interphase fluorescence in situ hybridization or karyotype. The most common abnormality is del6q, but it has no proven prognostic effect.37 Mutations/deletions of p53 are associated with poor prognosis,38 but are uncommon and rarely evaluated in clinical practice.
Therapies for WM
WM is a rare disease, and few randomized trials have been conducted. No approved drugs or combination existed for WM until recently, when the US Food and Drug Administration and European Medicines Agency approved ibrutinib. Most data come from phase 2 studies and cross-trial comparisons; there are very few studies directly comparing different regimens (Table 5).
Contemporary regimens used for the therapy of WM
| Regimen | Phase/disease setting/number of patients | Regimen details and duration of therapy | ORR | Major RR (at least PR) | IgM flare | Time to response | PFS | OS | Comments |
|---|---|---|---|---|---|---|---|---|---|
| Rituximab64 | Phase 3, untreated and pretreated (all rituximab sensitive; N = 75) | Rituximab intravenously 375 mg/m2 on day 1 of weeks 1-4 and 17-20; 8 infusion total | 48% | 33% | 47% | NR | 20.3 months | 3-year OS: 90% | IgM flare common |
| DRC43,99 | Phase 2, untreated (N = 72) | 6 cycles | 83% | 74% | 32% | 4.1 months | 35 months | 95 months | MDS ∼1% DLBCL ∼3% |
| BR44 | Phase 3 (subanalysis), untreated (N = 22) | Bendamustine intravenously 90 mg/m2 days 1 and 2; rituximab intravenously 375 mg/m2 on day 1; up to six 28-day cycles | 95% | 95% | NR | NR | 69 months | 90.4% at 5 years | Subgroup analysis of a larger study comparing BR with R-CHOP |
| BR45 | Retrospective, untreated (N = 69) | Bendamustine intravenously 90 mg/m2 days 1 and 2; rituximab intravenously 375 mg/m2 on day 1; up to six 28-day cycles; 56% completed 6 cycles | 97% | 96% | NR | <3 months | 2-year PFS: 87% | 2-year PFS: 97% | In 30% bendamustine, reduction to ≤70 mg/m2 |
| BR46 | Retrospective, pretreated (N = 71) | Bendamustine intravenously 50-90 mg/m2 days 1 and 2; rituximab intravenously 375 mg/m2 on day 1; up to six 28-day cycles; 66% completed 6 cycles | 80.2% | 74.6% | NR | 3 months | NR | NR | 37% received bendamustine ≤70 mg/m2 |
| BDR48,53 | Phase 2, untreated (N = 59) | Cycle 1: bortezomib 1.3 mg/m2 intravenously days 1, 4, 8, and 11 (21-day cycle); cycles 2-5: bortezomib 1.6 mg/m2 intravenously days 1, 8, 15, and 22 every 35 days; cycles 2 and 5: dexamethasone 40 mg; rituximab intravenously 375 mg/m2 (total 8 infusions of rituximab); 5 cycles | 85% | 68% | 11% | 3 months | 43 months | 66% at 8 years | No MDS DLBCL in 5% |
| BDR51,54 | Phase 2, untreated (N = 23) | Cycles 1-4: bortezomib intravenously 1.3 mg/m2; dexamethasone 40 mg on days 1, 4, 8, and 11; rituximab 375 mg/m2 day 11; cycles 5-8: as above, given 3 months apart; 4 + 4 cycles | 96% | 91% | 9% | 1.4 months | 57% at 5 years | 95% at 5 years | Neuropathy leading to discontinuation of bortezomib 60% |
| VR49 | Phase 2, untreated (N = 26) | Cycles 1-6: bortezomib intravenously 1.6 mg/m2 weekly days 1, 8, and 15, every 28 days; cycles 1 and 4: rituximab intravenously 375 mg/m2 weekly, 6 cycles | 88% | 66% | 31% | 3.7 months | 37 months | 94% at 5 years | Weekly bortezomib |
| VR50 | Phase 2, pretreated (N = 37) | Cycles 1-6: bortezomib intravenously 1.6 mg/m2 weekly, days 1, 8, and 15, every 28 days; cycles 1 and 4: rituximab intravenously 375 mg/m2 weekly, 6 cycles | 81% | 51% | 22% | 2 months | 19 months | 65% at 5 years | Weekly bortezomib |
| IRD56 | Phase 2, untreated (N = 26) | Cycles 1-2: ixazomib, by mouth 4 mg, days 1, 8, and 15; dexamethasone, by mouth or intravenously 20 mg, days 1, 8, and 15 every 4 weeks; cycles 3-6: ixazomib, by mouth 4 mg, days 1, 8, and 15; dexamethasone, by mouth or intravenously 20 mg, days 1, 8, and 15; rituximab intravenously 375 mg/m2, on day 1, every-4-week maintenance therapy: as above every 8 weeks for 6 cycles; 6 + 6 cycles | 96% | 77% | 8% | 2 months (8 wk) | 73% at 22 months | 100% at 22 months | Response was slower among those with CXCR4WHIM |
| FCR58 | Phase 2, pretreated (N = 40) | Rituximab intravenously 375 mg/m2 day 1; fludarabine intravenously 25 mg/m2 days 2-4; cyclophosphamide intravenously 250 mg/m2 days 2-4; 6 cycles | 80% | 80% | 0% | 3 months | Not reached at 55 months | NR | MDS/AML, 5%; transformation, 2.5%; infectious deaths 5% |
| FCR59 | Retrospective untreated (N = 25); pretreated (N = 57) | Rituximab intravenously 375 mg/m2 day 1; fludarabine by mouth 40 mg/m2 days 1-3; cyclophosphamide by mouth 250 mg/m2 days 1-3 in 28-day cycles; 6 cycles | 85.4% | 64.6% | 0 | NR (time to best response 10.8 months) | 67% at 48 months | 90% at 3 years | MDS/AML (N = 2); transformation (N = 3); long-lasting cytopenias (N = 19) late improved responses in 25 patients |
| Ibrutinib62 | Phase 2 pretreated (N= 63) | Ibrutinib by mouth 420 mg/ continuous | 90.5% | 73% | 0% | 1 months | 60% at 5 years | 87% at 5 years | Response and PFS lower in CXCR4WHIM and MYD88WT |
| Ibrutinib63 | Phase 3 (companion study), pretreated (N = 31; all rituximab refractory) | Ibrutinib by mouth 420 mg/day continuous | 90% | 71% | 0% | 1 months | 86% at 18 months | 97% at 18 months | Response and PFS similar in CXCR4WHIM, but slower |
| Ibrutinib65 | Phase 2, untreated (N = 30) | Ibrutinib by mouth 420 mg/day continuous | 100% | 83% | 0% | 1 months | 92% at 18 months | 100% at 18 months | Major responses (94% vs 71%) and VGPRs (31% vs 7%) higher and time to major response shorter (1.8 vs 7.3 months) in patients with CXCR4WT vs CXCR4WHIM |
| Ibrutinib-Rituximab64 | Phase 3, untreated and pretreated (all rituximab sensitive; N = 75) | Ibrutinib by mouth 420 mg/day, continuous (given before rituximab infusion), rituximab intravenously 375 mg/m2 on day 1 of weeks 1-4 and 17-20; 8 infusion total | 92% | 72% | 8% | 1 months | 82% at 30 months | 94% at 30 months | Randomized study, PFS and response not affected by MY88 and CXCR4 mutation status |
| Venetoclax93 | Phase 2, pretreated (N=30); ibrutinib exposed (N=15) | Venetoclax by mouth 200 mg/day, increased to 800 mg for 2 years | 87% | 74%* | NR | NR | NR | NR | Effective in ibrutinib-exposed patients |
| Regimen | Phase/disease setting/number of patients | Regimen details and duration of therapy | ORR | Major RR (at least PR) | IgM flare | Time to response | PFS | OS | Comments |
|---|---|---|---|---|---|---|---|---|---|
| Rituximab64 | Phase 3, untreated and pretreated (all rituximab sensitive; N = 75) | Rituximab intravenously 375 mg/m2 on day 1 of weeks 1-4 and 17-20; 8 infusion total | 48% | 33% | 47% | NR | 20.3 months | 3-year OS: 90% | IgM flare common |
| DRC43,99 | Phase 2, untreated (N = 72) | 6 cycles | 83% | 74% | 32% | 4.1 months | 35 months | 95 months | MDS ∼1% DLBCL ∼3% |
| BR44 | Phase 3 (subanalysis), untreated (N = 22) | Bendamustine intravenously 90 mg/m2 days 1 and 2; rituximab intravenously 375 mg/m2 on day 1; up to six 28-day cycles | 95% | 95% | NR | NR | 69 months | 90.4% at 5 years | Subgroup analysis of a larger study comparing BR with R-CHOP |
| BR45 | Retrospective, untreated (N = 69) | Bendamustine intravenously 90 mg/m2 days 1 and 2; rituximab intravenously 375 mg/m2 on day 1; up to six 28-day cycles; 56% completed 6 cycles | 97% | 96% | NR | <3 months | 2-year PFS: 87% | 2-year PFS: 97% | In 30% bendamustine, reduction to ≤70 mg/m2 |
| BR46 | Retrospective, pretreated (N = 71) | Bendamustine intravenously 50-90 mg/m2 days 1 and 2; rituximab intravenously 375 mg/m2 on day 1; up to six 28-day cycles; 66% completed 6 cycles | 80.2% | 74.6% | NR | 3 months | NR | NR | 37% received bendamustine ≤70 mg/m2 |
| BDR48,53 | Phase 2, untreated (N = 59) | Cycle 1: bortezomib 1.3 mg/m2 intravenously days 1, 4, 8, and 11 (21-day cycle); cycles 2-5: bortezomib 1.6 mg/m2 intravenously days 1, 8, 15, and 22 every 35 days; cycles 2 and 5: dexamethasone 40 mg; rituximab intravenously 375 mg/m2 (total 8 infusions of rituximab); 5 cycles | 85% | 68% | 11% | 3 months | 43 months | 66% at 8 years | No MDS DLBCL in 5% |
| BDR51,54 | Phase 2, untreated (N = 23) | Cycles 1-4: bortezomib intravenously 1.3 mg/m2; dexamethasone 40 mg on days 1, 4, 8, and 11; rituximab 375 mg/m2 day 11; cycles 5-8: as above, given 3 months apart; 4 + 4 cycles | 96% | 91% | 9% | 1.4 months | 57% at 5 years | 95% at 5 years | Neuropathy leading to discontinuation of bortezomib 60% |
| VR49 | Phase 2, untreated (N = 26) | Cycles 1-6: bortezomib intravenously 1.6 mg/m2 weekly days 1, 8, and 15, every 28 days; cycles 1 and 4: rituximab intravenously 375 mg/m2 weekly, 6 cycles | 88% | 66% | 31% | 3.7 months | 37 months | 94% at 5 years | Weekly bortezomib |
| VR50 | Phase 2, pretreated (N = 37) | Cycles 1-6: bortezomib intravenously 1.6 mg/m2 weekly, days 1, 8, and 15, every 28 days; cycles 1 and 4: rituximab intravenously 375 mg/m2 weekly, 6 cycles | 81% | 51% | 22% | 2 months | 19 months | 65% at 5 years | Weekly bortezomib |
| IRD56 | Phase 2, untreated (N = 26) | Cycles 1-2: ixazomib, by mouth 4 mg, days 1, 8, and 15; dexamethasone, by mouth or intravenously 20 mg, days 1, 8, and 15 every 4 weeks; cycles 3-6: ixazomib, by mouth 4 mg, days 1, 8, and 15; dexamethasone, by mouth or intravenously 20 mg, days 1, 8, and 15; rituximab intravenously 375 mg/m2, on day 1, every-4-week maintenance therapy: as above every 8 weeks for 6 cycles; 6 + 6 cycles | 96% | 77% | 8% | 2 months (8 wk) | 73% at 22 months | 100% at 22 months | Response was slower among those with CXCR4WHIM |
| FCR58 | Phase 2, pretreated (N = 40) | Rituximab intravenously 375 mg/m2 day 1; fludarabine intravenously 25 mg/m2 days 2-4; cyclophosphamide intravenously 250 mg/m2 days 2-4; 6 cycles | 80% | 80% | 0% | 3 months | Not reached at 55 months | NR | MDS/AML, 5%; transformation, 2.5%; infectious deaths 5% |
| FCR59 | Retrospective untreated (N = 25); pretreated (N = 57) | Rituximab intravenously 375 mg/m2 day 1; fludarabine by mouth 40 mg/m2 days 1-3; cyclophosphamide by mouth 250 mg/m2 days 1-3 in 28-day cycles; 6 cycles | 85.4% | 64.6% | 0 | NR (time to best response 10.8 months) | 67% at 48 months | 90% at 3 years | MDS/AML (N = 2); transformation (N = 3); long-lasting cytopenias (N = 19) late improved responses in 25 patients |
| Ibrutinib62 | Phase 2 pretreated (N= 63) | Ibrutinib by mouth 420 mg/ continuous | 90.5% | 73% | 0% | 1 months | 60% at 5 years | 87% at 5 years | Response and PFS lower in CXCR4WHIM and MYD88WT |
| Ibrutinib63 | Phase 3 (companion study), pretreated (N = 31; all rituximab refractory) | Ibrutinib by mouth 420 mg/day continuous | 90% | 71% | 0% | 1 months | 86% at 18 months | 97% at 18 months | Response and PFS similar in CXCR4WHIM, but slower |
| Ibrutinib65 | Phase 2, untreated (N = 30) | Ibrutinib by mouth 420 mg/day continuous | 100% | 83% | 0% | 1 months | 92% at 18 months | 100% at 18 months | Major responses (94% vs 71%) and VGPRs (31% vs 7%) higher and time to major response shorter (1.8 vs 7.3 months) in patients with CXCR4WT vs CXCR4WHIM |
| Ibrutinib-Rituximab64 | Phase 3, untreated and pretreated (all rituximab sensitive; N = 75) | Ibrutinib by mouth 420 mg/day, continuous (given before rituximab infusion), rituximab intravenously 375 mg/m2 on day 1 of weeks 1-4 and 17-20; 8 infusion total | 92% | 72% | 8% | 1 months | 82% at 30 months | 94% at 30 months | Randomized study, PFS and response not affected by MY88 and CXCR4 mutation status |
| Venetoclax93 | Phase 2, pretreated (N=30); ibrutinib exposed (N=15) | Venetoclax by mouth 200 mg/day, increased to 800 mg for 2 years | 87% | 74%* | NR | NR | NR | NR | Effective in ibrutinib-exposed patients |
MDS, myelodysplastic syndrome; PR, partial response; R-CHOP, rituximab-cyclophosphamide, doxorubicin, vincristine, prednisone; VGPR, very good partial response.
87% in ibrutinib naive and 60% in ibrutinib exposed.
The most rapidly acting therapy, whenever immediate IgM reduction is required (such as for hyperviscosity, symptomatic cryoglobulinemia, severe hemolysis resulting from cold agglutinin disease, etc) is plasmapheresis (Figures 1 and 2); blood warmers should be considered during apheresis if cryoglobulins are present.39 After 2 to 3 plasmapheresis sessions, IgM levels can be reduced significantly, but the effect is only transient, and systemic therapy is required.

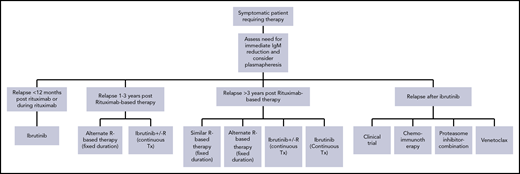
Management of patients that relapse after rituximab-based therapy. For patients who relapse after ibrutinib or discontinue ibrutinib for reasons such as toxicity, there are limited data, and venetoclax or anti-CD20 monoclonal antibody with chemotherapy or proteasome inhibitors may be considered, depending on the availability of venetoclax and other drugs.
Management of patients that relapse after rituximab-based therapy. For patients who relapse after ibrutinib or discontinue ibrutinib for reasons such as toxicity, there are limited data, and venetoclax or anti-CD20 monoclonal antibody with chemotherapy or proteasome inhibitors may be considered, depending on the availability of venetoclax and other drugs.
Rituximab-based combinations are the most commonly used systemic therapies.40 Rituximab monotherapy is slow to act; an extended regimen (8 infusions) has better activity. A transient increase of serum IgM (IgM flare) is common, occurring in 30% to 80% of patients treated with rituximab or other anti-CD20 monoclonal antibodies, and may exacerbate IgM-related complications.41,42 Rituximab with oral or intravenous cyclophosphamide and dexamethasone (DRC, for 6 cycles) remains a commonly used regimen with low short- and long-term toxicity, but responses may delay for months, complete responses are rare, and IgM flare is common.43 Bendamustine with rituximab (BR, for 4-6 cycles) is active (response rates ∼90%, including some complete responses) and with long-lasting responses.44,45 In older patients, bendamustine dose reductions are required, and up to 4 cycles of BR may be sufficient, as hematologic toxicity and infectious complications are common.45-47 There are limited data on IgM flare in patients treated with BR.
Bortezomib, which has shown activity in several phase 2 studies, either alone or in combination with rituximab (VR or BDR) in either newly diagnosed or relapsed patients,48-51 does not cause IgM flare and can rapidly reduce IgM levels, but marrow clearance may lag behind when used alone.52 Today, bortezomib is administered subcutaneously and in weekly intervals49,50,53 to reduce neurotoxicity. These fixed-duration alkylator-free regimens, even without maintenance, offer prolonged progression free survival (PFS).48,54 A randomized prospective study (ECWM-1) comparing DRC vs DRC with bortezomib in previously untreated symptomatic patients has completed accrual. The nonneurotoxic proteasome inhibitor carfilzomib has been tested in combination with rituximab in a small trial, but carfilzomib may be associated with a risk for cardiotoxicity.55 Ixazomib, an oral proteasome inhibitor structurally similar to bortezomib, could be useful in WM, but the data are immature; it was evaluated in combination with rituximab and dexamethasone in previously untreated patients with response rates similar to bortezomib-rituximab,56 whereas another study evaluates ixazomib/dexamethasone with rituximab in patients with relapsed WM.
Combinations of nucleoside analogs with rituximab57-59 are very active, but have significant short- and long-term toxicity and are not primary options.58-60 Combinations with intensive chemotherapy, such as rituximab-cyclophosphamide, doxorubicin, vincristine, and prednisone, are only considered if transformation to DLBCL occurs. Single-agent chemotherapy is rarely considered, but single-agent oral fludarabine is more effective than chlorambucil.61
Ibrutinib is the most active single agent in WM,62,63 but complete responses are rare either as monotherapy or when combined with rituximab.64 In the prospective randomized iNNOVATE trial, ibrutinib with rituximab was compared with extended-schedule rituximab with placebo in previously untreated or pretreated WM.64 The ibrutinib-rituximab combination reduced the risk for disease progression by 80%. In previously untreated patients, ibrutinib monotherapy resulted in high response rates (no complete responses); these occurred slower in those carrying CXCR4WHIM, but the follow-up is still short.65 Ibrutinib is active in both rituximab-sensitive and rituximab-refractory patients.62,63 In the initial phase 2 study, 5-year PFS was 60% and 5-year overall survival (OS) was 87%, but responses and PFS were better in patients with the MYD88L265P/CXCR4WT than the MYD88L265P/CXCR4WHIM genotype; among the few patients with MYD88WT, the responses were minor and PFS short.14 In the iNNOVATE study, ibrutinib/rituximab-treated patients harboring CXCR4WHIM had lower very good partial response rates and shorter PFS at 3 years, but those with MYD88WT had similar outcomes to MYD88L265P. Ibrutinib is associated with a risk for atrial fibrillation (in ∼10% to 12% of patients)64 and hemorrhages (usually minor, but risk increases with anticoagulants), has several interactions with commonly used drugs (antibiotics, antiarrhythmics, etc), and requires continuous uninterrupted therapy. An ibrutinib withdrawal syndrome can occur, characterized by B symptoms and IgM rebound in ∼20% of patients that interrupt ibrutinib for unrelated reasons66,67 ; however, most patients recover on reinitiation and eventually reachieve IgM response.67 Two new BTK inhibitors are tested in WM in phase 2 (acalabrutinib) or phase 3 (zanabrutinib compared with ibrutinib) studies.
Choosing primary therapy
Patients with WM may present with a variety of symptoms and complications (Table 1), sometimes requiring immediate disease control. The goal of therapy is to control symptoms and reduce tumor burden; a complete response is difficult to achieve with current therapies. In patients presenting with high tumor bulk (extensive organomegaly/lymphadenopathy, BM infiltration, high b2-microglobulin, elevated LDH, B symptoms), rapidly acting regimens are preferred. If organomegaly, lymphadenopathy, and B symptoms predominate, then BR or ibrutinib/rituximab may be preferable. In patients with cytopenias, bortezomib/rituximab (±dexamethasone) or ibrutinib/rituximab may be preferred over BR because of lower myelotoxicity. For patients presenting with or at risk for hyperviscosity, severe cryoglobulinemia or cold agglutinin disease, plasmapheresis should be considered. A regimen containing bortezomib, including a short induction before starting rituximab53 or ibrutinib, can rapidly reduce IgM levels and risk for IgM flare.64 When rapid reduction of toxic IgM is needed (as in AL amyloidosis, cryoglobulinemia, cold agglutinin disease73 ), regimens such as BR74 or BDR53 are better options; ibrutinib may be less preferable in patients with cardiac amyloidosis because of the risk for atrial fibrillation (AF).75 In patients not in need for immediate disease control (low tumor bulk, or with mild cytopenias or mild symptoms), DRC, which has low toxicity, provides a safe, low-cost, fixed-duration option; BR is very active but may be more toxic (Figure 1).
Not all patients with IgM-related neuropathy require immediate therapy, and those with mild symptoms and no other reason for treatment should not start therapy, but be followed closely.24 For patients requiring therapy, rituximab monotherapy is not very effective,76,77 and may exacerbate symptoms resulting from IgM flare. DRC may be preferable to rituximab alone, BR may be toxic, and there is limited experience; there is some experience with FCR,78 but it is very toxic. Ibrutinib may be a reasonable choice in selected patients with IgM-related neuropathy62 ; bortezomib is associated with a risk for neurotoxicity. Bing-Neel syndrome is a rare manifestation of WM resulting from the infiltration of the central nervous system by malignant lymphoplasmacytic cells. Treatments may include high-dose methotrexate protocols; fludarabine has shown activity,79 but ibrutinib and other BTKis, penetrate the blood-brain barrier, have shown promising activity,80 and may be a primary option.
Age critically influences treatment selection. Short-term toxicity is a major concern in the elderly because of frailty, whereas WM-unrelated mortality is significant2 ; symptom and disease control should be the initial goal for most. Less toxic combinations such as DRC are well tolerated; BR requires dose reductions, and BDR carries a risk for neuropathy. Ibrutinib (monotherapy or with rituximab) is very active, but the risk for AF is higher in the elderly,81,82 and treatment may be challenging in those with cardiac comorbidities or in need of anticoagulation or double-antiplatelet therapy or in those receiving drugs that interact with ibrutinib. In younger patients, deeper responses may be the goal; short-term toxicity is better tolerated, but long-term toxicity (secondary malignancies, MDS, disease transformation resulting from exposure to alkylators, nucleoside analogs, or prolonged immunosuppression) is of concern; thus, alkylator-free regimens such as BDR may be preferable. BR and DRC also have relatively low long-term toxicity. Ibrutinib has not been associated with MDS risk or disease transformation, but long-term effects are not fully known.
After the introduction of ibrutinib, the concept of continuous vs fixed-duration therapy has emerged. Both approaches have pros and cons. Ibrutinib induces rapid responses, but therapy continues uninterrupted for years (unknown for how long); the previously described toxicities may complicate therapy in elderly frail patients, and perhaps some currently unknown long-term risks may exist, whereas the financial burden is high. Approaches to limit the need for continuous therapy by combining BTKs with other drugs (venetoclax, proteasome inhibitors, etc) are ongoing. Rituximab-based chemoimmunotherapy is given for a fixed, limited duration, usually a few months, with some patients also receiving fixed-duration maintenance (Table 5), with a relatively long treatment-free interval and well-recognized short- and long-term risks.
How I treat
Case 1
About 18 months after initial diagnosis, the patient complained of worsening fatigue and low-grade fever. IgM is 4.4 g/dL, but his hemoglobin is 11.8 g/dL. Per IPSSWM, the patient is low risk, and per rIPSSWM, he is low risk (has a score of 1).
Our primary consideration is to assess the need for rapid disease control and patient’s age. For most patients, our primary choice is a rituximab-based, fixed-duration therapy, offering a treatment-free interval of several years (Table 5; Figure 1). This young patient started therapy with BDR, achieved a rapid response, and completed 6 cycles without maintenance. Although maintenance rituximab could provide some clinical benefit, according to retrospective data,83 it cannot be recommended because of the lack of prospective data in WM; the results of the MAINTAIN study, which compares 2 years of rituximab maintenance vs no maintenance after BR, are awaited. An approach based on ibrutinib (monotherapy62 or with rituximab64 ) could also be considered.
Case 2
An 80-year-old woman with a history of hypertension, paroxysmal atrial fibrillation, and depression was referred for normocytic anemia (hemoglobin of 7.5 g/dL) and weight loss. SPEP revealed a monoclonal IgM(κ) of 6.2 g/dL. A BM biopsy showed extensive infiltration by monoclonal lymphoplasmacytes and plasma cells harboring MYD88L265P.
Along with disease characteristics, the age, comorbidities, patient’s preferences, and drug availability are decisive factors for choice of therapy. This elderly patient declined chemoimmunotherapy and started receiving ibrutinib, achieving a rapid IgM reduction, and her hemoglobin increased. She developed AF, and as a result of a severe infection, temporarily discontinued ibrutinib: IgM increased by >50% within 1.5 month, but when she restarted ibrutinib, IgM dropped again. She continues ibrutinib with anticoagulation (with apixaban). Concurrent use of ibrutinib with anticoagulation is challenging, and thrombotic risk should be weighed against bleeding risk (with tools such as CHA2DS2-VASc and HAS-BLED) and potential alternative treatment options. There are data from ibrutinib-treated patients with chronic lymphocytic leukemia (CLL) and expert opinions84-86 regarding the management of such patients, but often the recommendations are conflicting.87 In our practice, we prefer to use apixaban or dabigatran (which also has an antidote available) at lower doses.
Case 3
A 76-year-old man with a history of heart failure resulting from coronary artery disease and valvular heart disease was admitted for dyspnea. Anemia (hemoglobin 8.3 g/dL), thrombocytopenia (platelet counts 88 × 109/L), oronasal bleeding, high serum total protein (13.4 g/dL), and low serum albumin (2.6 g/dL) were noted. SPEP revealed monoclonal IgM(κ) (∼8.1 g/dL). A BM biopsy revealed lymphoplasmacytic infiltration ∼90%. CT showed increased spleen size and multiple small lymph nodes in the mediastinum and abdomen.
For this patient, ibrutinib may not be the best option because of cardiac comorbidities. He received 2 plasmapheresis sessions to control hyperviscosity symptoms and started bortezomib. After 4 weekly bortezomib administrations, rituximab was added, and he has completed 4 cycles of BDR. Another option could also be BR at reduced doses.
Case 4
A 74-year-old woman was diagnosed with WM 7 years ago. She had anemia (hemoglobin, 9.8 g/dL) and 2.5 g/dL of IgM(κ), and BM had 80% LPL infiltration. She received 6 cycles of DRC, and IgM dropped to 0.9 g/dL and hemoglobin increased (12.6 g/dL). Eighteen months ago, a gradual increase of IgM was noted, and lately, she feels fatigued. Hemoglobin is 10.1 g/dL, and IgM is 2.4 g/dL.
The criteria to start therapy in newly diagnosed patients apply also in relapsing patients, but clinical judgment is important. For patients who relapse after a long remission after a rituximab-containing regimen (ie, at least 3 years, the median expected for regimens like DRC), a second attempt to achieve another prolonged remission with the initial or a different rituximab-based regimen (bendamustine instead of cyclophosphamide88 or a proteasome inhibitor49 ) is reasonable, but no prospective randomized data exist (Table 5; Figure 2). Combinations with a nucleoside analog57,58 are effective, but carry significant risk for myelotoxicity and MDS,58,89 and we rarely use them. Ibrutinib alone62 or in combination with rituximab64 is very active. There is no direct comparison of Ibrutinib/rituximab with ibrutinib alone in WM. Data from CLL indicate that probably there is no difference, but according to iNNOVATE data, in patients with MYD88WT, ibrutinib with rituximab may be preferable to ibrutinib alone,64 as these patients seem to have similar outcomes to those bearing MYD88L265P. We do not choose therapy on the basis of CXCR4 status; however, for patients with unknown MYD88 mutational status, we recommend testing before initiation of ibrutinib therapy. For patient 4, repeating DRC43 or starting BR was discussed. She received BR (bendamustine 70 mg/m2) for 4 cycles and achieved a PR, and her hemoglobin improved. Her IgM continued to drop for 9 months after BR completion, and remained in remission for 28 months.
Case 1
The patient relapsed 14 months after completion of primary therapy with BDR. Repeat genotyping showed MYD88L265P and no CXCR4WHIM.
In patients who relapse more than 12 months after the last rituximab dose, but in whom the duration of remission was less than 3 years, ibrutinib (±rituximab) may be the most active therapy. Another rituximab-based regimen, different than the one previously used, may be considered as well. Although it has not been directly compared with regimens such as BR or BDR, ibrutinib is probably more effective in this patient population. For this patient, potential options included a combination of chemotherapy with rituximab or ibrutinib. Because of his young age, avoiding exposure to chemotherapy was felt to be important, and the patient started ibrutinib with excellent tolerability and response.
If disease progresses during rituximab therapy or less than 12 months after last rituximab dose, then WM is considered rituximab resistant. However, IgM flare may falsely give the impression of disease progression during rituximab therapy. Additional tests and clinical evaluation (eg, anemia improving and resolution of B symptoms) help differentiate flare from true progression. Also, delayed IgM responses after rituximab-based therapy can occur even months after completion of therapy. It is not clear whether progression during or shortly after rituximab maintenance (given every 8-12 weeks) also represents true resistance. For patients with rituximab-resistant disease, ibrutinib monotherapy is the preferred and most active therapy.63 Single-agent bortezomib has shown activity in small studies,52,90 but results are inferior to those of ibrutinib. Some encouraging results from single-agent carfilzomib should be viewed with caution because of the small numbers of patients.91 A different rituximab-based combination in a rituximab-refractory patient may have efficacy, but specific data are limited. Ofatumumab, another anti-CD20 monoclonal antibody, has some efficacy, but responses are short-lasting in rituximab-refractory patients.60,92
An emerging challenge is the management of patients progressing during ibrutinib therapy or those discontinuing because of toxicity. In a recent phase 2 study, among 30 patients with relapsed/refractory WM (15 had prior ibrutinib), single-agent venetoclax induced responses in 93% of the patients, which, however, were more frequent and deeper in ibrutinib-naive patients.93 Venetoclax has been approved for the treatment of CLL and could be an option for patients failing ibrutinib. Many patients failing ibrutinib can be salvaged with rituximab-based chemoimmunotherapy,67 but their outcome is often poor; participation in clinical trials with new agents is the best option.
High-dose therapy with autologous stem cell transplantation may have a role for the treatment of young patients with chemosensitive disease or those with an early relapse and a clinically aggressive course.94 Given the toxicity of high-dose therapy, other options may be more attractive. In patients who develop disease transformation to high-grade lymphoma, high-dose therapy may be part of the treatment. The role of allogeneic transplantation remains limited, and could be considered only in carefully selected patients failing BTK inhibitors.95,96 CD19-directed CAR-T cell-based approaches are in clinical development, with very few WM patients treated yet.
Most patients with WM will have a prolonged disease course with multiple lines of therapy, and may struggle with various complications. Patients with WM are at risk for infections; hypogammaglobulinemia is common and persists despite response to treatment, but is not associated with the incidence of recurrent infections.19 Vaccinations are recommended; use of intravenous immunoglobulin is not recommended unless frequent severe infections occur. Second primary malignancies (solid tumors, skin cancers, and myelodysplasia), as well as transformation to aggressive lymphomas, can occur. A significant proportion of previously untreated patients with WM already harbor evidence of clonal hematopoiesis of indeterminate potential.97 Physicians should educate their patients to adhere to standard screening for solid tumors, and appropriately evaluate anemia or cytopenias not related to disease progression.
Conclusions
Many options that fit the needs of different patients with WM are available. The major challenge is the development of active, low-toxicity combinations that will provide a high probability of complete responses, potentially with a fixed duration of therapy. New treatments are emerging, but available therapies can be further optimized. Inclusion in clinical trials offers the best opportunity for patients to receive new therapies, and for most patients, this would be our preferred choice. Despite recent advances, we need deeper understanding of the disease and international collaboration in clinical trials to improve therapy.
Acknowledgment
The authors thank Maria Gavriatopoulou for contributing in data analysis and interpretation and the critical review of the manuscript.
Authorship
Contribution: M.A.D. and E.K. performed literature research and analysis and authored the manuscript.
Conflict-of-interest disclosure: M.A.D. has received honoraria from Amgen, Janssen, Celgene, Takeda, and BMS. E.K. has received honoraria from Amgen, Genesis Pharma, Janssen, and Takeda.
Correspondence: Meletios A. Dimopoulos, 80 Vas Sofias Ave and 7 Lourou Str, 11528, Athens, Greece; e-mail: mdimop@med.uoa.gr.
