

In this issue of Blood, eloquently describe the oncogenic interplay between the maternal embryonic leucine zipper kinase (MELK) and the enhancer of zeste homolog 2 (EZH2) in extranodal natural killer/T-cell lymphoma (NKTL) through the phosphorylation-dependent loss of ubiquitination. Their work significantly enhances our understanding of the pathophysiology of extranodal NKTL and provides additional evidence for targeting this enzymatic pathway in mature NK/T-cell malignancies.1
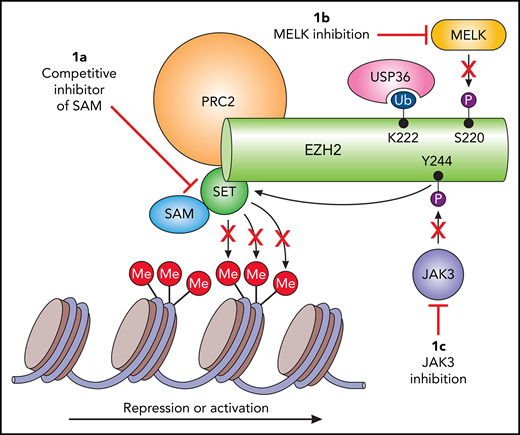
Potential targets of EZH2 in extranodal NKTL. (A) The majority of the currently investigated EZH2 small-molecule inhibitors hamper methylation of H3K27, resulting in decreased transcription repression. However, this strategy may not prove beneficial in extranodal NKTL. (B) MELK-mediated phosphorylation results in deubiquitination by USP36 and thus decreased degradation, and (C) illustrates another oncogenic mechanism of EZH2 in extranodal NKTL through JAK3 phosphorylation and subsequent transcription activation. This indicates that both JAK3 and MELK are potential targets for arresting EZH2 oncogenesis in extranodal NKTL. SET, Su(var)3-9, enhancer-of-zeste and trithorax.
Potential targets of EZH2 in extranodal NKTL. (A) The majority of the currently investigated EZH2 small-molecule inhibitors hamper methylation of H3K27, resulting in decreased transcription repression. However, this strategy may not prove beneficial in extranodal NKTL. (B) MELK-mediated phosphorylation results in deubiquitination by USP36 and thus decreased degradation, and (C) illustrates another oncogenic mechanism of EZH2 in extranodal NKTL through JAK3 phosphorylation and subsequent transcription activation. This indicates that both JAK3 and MELK are potential targets for arresting EZH2 oncogenesis in extranodal NKTL. SET, Su(var)3-9, enhancer-of-zeste and trithorax.
The Epstein-Barr virus (EBV)–driven NK/T-cell lymphoid disorders are a group of malignancies and lymphoproliferative diseases affecting both children and adults that exhibit strikingly heterogeneous clinical courses and outcomes.2 Arguably, the most well-known and researched of these NK/T-cell neoplasms is extranodal NKTL-nasal type, an aggressive disease with relatively poor survival.3
Extranodal NKTL represents less than 5% of all peripheral T-cell lymphomas in North America and Europe, but it accounts for up to one third of cases in East Asia.3 Roughly 70% of patients with extranodal NKTL present with localized or early-stage disease, most commonly as an infiltrative and destructive mass in the nasal cavity, or less frequently, as an extra-nasal mass involving the nasopharynx and upper aerodigestive tract which is associated with a worse prognosis and higher rates of disseminated disease.3 Survival for localized disease has improved significantly with the incorporation of radiation therapy into multiagent non-anthracycline–based chemotherapy, such as the dexamethasone, etoposide, ifosfamide, and carboplatin (DeVIC) regimen, with a 5-year progression-free survival (PFS) of 61% and an overall survival of 72%.4 Unfortunately, this regimen is associated with significant toxicities. Likewise, survival has improved in disseminated disease with the incorporation of l-asparaginase or pegylated asparaginase into multidrug regimens. The regimen of steroid, methotrexate, ifosfamide, l-asparaginase, and etoposide (SMILE) regimen resulted in a 1-year PFS of just over 50%, with similar results reported for other asparaginase-containing regimens.5,6 Despite these improvements in both localized and advanced-stage disease, a significant percentage of patients will relapse or exhibit refractory disease. Effective treatment options for these patients are limited, and survival is dismal. Targeting EZH2 with small-molecule inhibitors is showing promise in both B-cell and T-cell lymphomas (see figure) but is this also true for the extranodal NKTL?7
Histone modification through acetylation and methylation plays a crucial role in carcinogenesis. Specifically, for methylation, EZH2, and to a lesser extent EZH1, are necessary for survival of many hematologic and non-hematologic malignancies. The role of EZH2 and EZH1 in different tumors is undoubtedly multifaceted with aberrancies involving gain-of-function mutations, loss-of-function mutations, and protein overexpression.8 Remarkably, in B-cell lymphomas, wild-type EZH2 must be present for gain-of-function mutations to exhibit a survival benefit—a unique phenomenon among human cancers.9 Nevertheless, it was the discovery of gain-of-function mutations in B-cell lymphomas, mainly found in those of germinal center origin such as follicular lymphoma and a majority of diffuse large B-cell lymphomas, which led to the development of several EZH inhibitors.7,8
Li et al now describe an additional role of EZH2 in extranodal NKTL. Previous work from the same group described an alternative pathway whereby JAK3-mediated phosphorylation of EZH2 results in decreased methylation of histone H3 on Lys27 (H3K27). In other words, this site-specific phosphorylation transforms EZH2 into a transcriptional activator rather than a transcriptional repressor, the classic downstream function of EZH2 mutations found in other nodal lymphomas (see figure). Consequently, anti-tumor response in vitro with several EZH inhibitors that inhibit methyltransferase activity through S-adenosyl-methionine (SAM) competitive inhibition or inhibit other proteins of the polycomb repressive complex 2 (PRC2) were not effective.10 The Li et al group now report the discovery of the pathogenic link between EZH2 and MELK. The authors begin with the discovery that MELK and EZH2 are concordantly and significantly overexpressed in extranodal NKTL patient samples when compared with healthy NK cells. Both therapeutic inhibition of MELK and MELK knockdown led to decreased EZH protein levels, but not messenger RNA expression, suggesting that MELK is involved in EZH2 protein stability and/or function. Li et al later identified a unique site-specific MELK-mediated phosphorylation at S220 of EZH2, which required the addition of recombinant MELK to EZH2 for phosphorylation. At the same time, they found K222 on EZH2 to be the exclusive site for ubiquitination. Given the close proximity of K222 to S220, it was speculated that phosphorylation of S220 led to deubiquitination of K222 and thus conservation of the EZH2 protein. USP36 was observed to be the deubiquitinase responsible for maintaining a ubiquitin-free pocket, thereby allowing for ongoing phosphorylation at S220 driven by MELK. Finally, the authors describe increased sensitivity of extranodal NKTL to the proteasome inhibitor (PI) bortezomib in the absence of functional MELK, but this was not seen with the second-generation PI carfilzomib.
In summary, Li et al provide us with an additional piece to the puzzle of the oncogenesis of extranodal NKTL. Although in vitro studies showed little benefit for methyltransferase-targeted EZH2 inhibitors in extranodal NKTL, this has not been confirmed in clinical trials. However, given the work by Li and colleagues, the addition of MELK or JAK3 inhibitors may prove useful and is worth further study in extranodal NKTL. We are just beginning to unlock the full potential of epigenetic manipulation for the treatment lymphoma. One example would be combining EZH pathway inhibitors with other epigenetic modifiers such as the histone deacetylase inhibitors. This and other combinations warrant further investigation, particularly in EBV-driven lymphomas such as extranodal NKTL.
Conflict-of-interest disclosure: The authors declare no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal