

In this issue of Blood, demonstrate that the low factor V (FV) levels in patients with combined deficiency of FV and FVIII (F5F8D) may in fact be beneficial for the patients and ameliorate their bleeding tendency.1 This report adds to the growing list of observations that FV not only is procoagulant but also that it works as an anticoagulant. The paper brings new understanding of the basic pathology of F5F8D and also lays the foundation for a change in the treatment strategy of the disease.
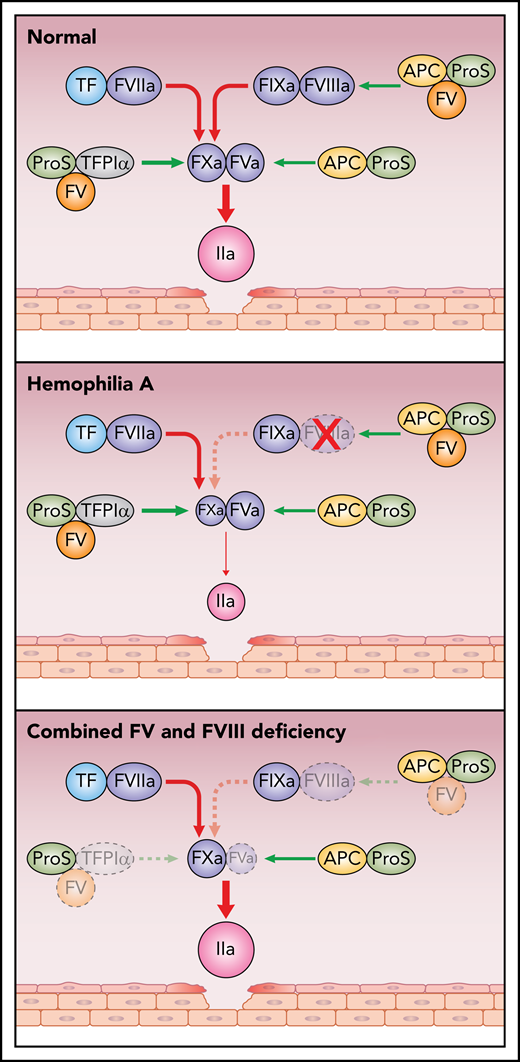
Simplified scheme of pro- and anticoagulant reactions in health, in hemophilia A, and in combined FV and FVIII deficiency. Damage to the vessel wall results in activation of coagulation via FVIIaTF. FVIIaTF and FIXaFVIIIa activate FX; FXa then binds to FVa and activates prothrombin to thrombin. The anticoagulant functions of FV include being a carrier to TFPIα and a synergistic cofactor with protein S (ProS) to TFPIα as well as to APC. In health, the pro- and anticoagulant mechanisms are in balance. In contrast, in hemophilia A, the balance is shifted to anticoagulation with little thrombin generated. In F5F8D, because the anticoagulant functions of FV, TFPIα, and protein S are deficient, the low levels of FV and FVIII are sufficient to efficiently generate thrombin. Red arrows, procoagulant reactions; green arrows, anticoagulant reactions.
Simplified scheme of pro- and anticoagulant reactions in health, in hemophilia A, and in combined FV and FVIII deficiency. Damage to the vessel wall results in activation of coagulation via FVIIaTF. FVIIaTF and FIXaFVIIIa activate FX; FXa then binds to FVa and activates prothrombin to thrombin. The anticoagulant functions of FV include being a carrier to TFPIα and a synergistic cofactor with protein S (ProS) to TFPIα as well as to APC. In health, the pro- and anticoagulant mechanisms are in balance. In contrast, in hemophilia A, the balance is shifted to anticoagulation with little thrombin generated. In F5F8D, because the anticoagulant functions of FV, TFPIα, and protein S are deficient, the low levels of FV and FVIII are sufficient to efficiently generate thrombin. Red arrows, procoagulant reactions; green arrows, anticoagulant reactions.
Blood coagulation is initiated by vascular damage, which exposes tissue factor (TF) to the circulating coagulation proteins.2,3 This initiates a series of proteolytic reactions that result in the formation of thrombin and a fibrin clot. Factor VIIa (FVIIa) bound to TF activates factors IX and X (FIX and FX), which team up with their respective activated cofactor, factor VIIIa and Va (FVIIIa and FVa) (see figure). FXaFVa activates prothrombin to generate the initial thrombin. Feedback from this reaction activates the system by converting the procofactors FVIII and FV to their active forms, and the system can now proceed at maximum speed.4 The role of FIXaFVIIIa is to provide amplification by generating more FXa. The cofactors FVa and FVIIIa are crucial as the enzymes FIXa and FXa have low intrinsic activities. This is illustrated by the severe bleeding phenotype affecting individuals with hemophilia A, due to defective or absent FVIII. In contrast, it has been puzzling that patients with FV deficiency generally have a mild bleeding phenotype. Patients with combined FVIII and FV deficiency also have a mild bleeding phenotype, and the report by Shao and colleagues provides a possible explanation by demonstrating that the low FV levels are associated with efficient thrombin generation. Reconstitution of FV to normal levels results in anticoagulation and decreased thrombin generation. This illustrates the importance of the anticoagulant functions of FV.
To date, 2 anticoagulant properties of FV have been identified (see figure). The discovery of activated protein C (APC) resistance, caused by the FVLeiden mutation, as a major risk factor for venous thrombosis led to the identification of FV as an APC cofactor functioning in synergy with the anticoagulant protein S in the degradation of FVIII in the FIXaFVIIIa complex.4,5 Loss of the APC cofactor activity due to the FVLeiden mutation is one of the mechanisms that generates a hypercoagulable state. The second anticoagulant function of FV is related to its interaction with full-length tissue factor pathway inhibitor (TFPIα).5 TFPIα, which regulates both the FVIIaTF complex and FXa, circulates bound to FV. As a consequence, individuals with FV deficiency have low TFPIα and therefore defective anticoagulation, which may be the explanation for the mild bleeding phenotype in FV deficiency.6 FV serves not only as a carrier of TFPIα in circulation but also as a synergistic TFPIα cofactor together with protein S in the inhibition of FXa.7,8 A minor splice isoform of FV, denoted FV-Short, which is particularly efficient as a TFPIα carrier and synergistic TFPIα cofactor with protein S, was discovered during elucidation of the pathogenic mechanism of the East Texas bleeding disorder.9
The low plasma levels of FV and FVIII in F5F8D result from defective secretion of FV and FVIII due to mutations in either LMAN1 (lectin mannose binding-1) or MCFD2 (multiple coagulation factor deficiency-2). The block in secretion is not complete, and therefore, the FV and FVIII plasma levels usually are 10% to 20% of normal. The associated bleeding phenotype is mild to moderate, and bleeding episodes have been treated on demand to increase FV and FVIII levels. Shao and colleagues have used an in vitro thrombin generation assay to investigate platelet-poor plasma from 6 patients with F5F8D. They made the surprising observation that thrombin generation in F5F8D was considerably higher than in that of a normal control. Addition of FVIII increased the thrombin generation further, whereas reconstitution of FV decreased thrombin generation. Because FV is known to carry TFPIα in plasma, and because FV deficiency is associated with low TFPIα, the authors measured TFPIα in the patients and found that the TFPIα plasma concentrations were equally low as those in severe FV deficiency. Subsequent reconstitution of TFPIα to the F5F8D patient plasma resulted in decreased thrombin generation. Platelet-rich plasma demonstrated a different picture with low thrombin generation in F5F8D patients. Addition of FV or TFPIα minimally affected thrombin generation, whereas FVIII addition greatly improved thrombin generation. Thus, in F5F8D patients, the thrombin generation patterns were very different in the presence or absence of platelets, which raises questions about the in vivo relevance in F5F8D patients of these findings. However, an important conclusion that can be drawn from both platelet-rich and platelet-poor plasma is that increase in FVIII results in increased thrombin generation. Increased FVIII levels can be achieved by infusion of 1-Deamino-8-DArgininVasoPressin (DDAVP), which induces secretion of FVIII and von Willebrand factor from endothelial cells but does not affect FV. DDAVP has been used in several cases of F5F8D, and the observation of Shao and colleagues provide a molecular basis for the rationale to use this treatment in F5F8D patients.
FV is a Janus-faced protein with both pro- and anticoagulant properties. The low FV levels in F5F8D may contribute to the mild phenotype. Based on the observation made by Shao and colleagues, it is tempting to speculate that selective inhibition of the anticoagulant properties of FV could be beneficial for hemophilia patients, but this may be difficult to achieve. However, several reports have demonstrated improved in vivo hemostasis in hemophilia after inhibition of different anticoagulant proteins (eg, antithrombin, TFPIα protein C, and protein S).10
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal