

Key Points
The JAK1/2 inhibitor ruxolitinib dampens inflammation in HLH via IFN-γ–dependent and –independent mechanisms.
Ruxolitinib exerts its beneficial effects in HLH by targeting T-cell and neutrophil activation and tissue infiltration.

Abstract
Hemophagocytic lymphohistiocytosis (HLH) is an often-fatal disorder characterized by the overactivation of T cells and macrophages that excessively produce proinflammatory cytokines, including interferon-γ (IFN-γ). Previously, we reported that the JAK inhibitor ruxolitinib dampens T-cell activation and lessens inflammation in a model of HLH in which perforin-deficient (Prf1−/−) mice are infected with lymphocytic choriomeningitis virus (LCMV). Ruxolitinib inhibits signaling downstream of IFN-γ, as well as several other JAK-dependent cytokines. As a consequence, it remained unclear whether ruxolitinib was exerting its beneficial effects in HLH by inhibiting IFN-γ signaling or by targeting signaling initiated by other proinflammatory cytokines. To address this question, we compared the effects of ruxolitinib with those obtained using an IFN-γ–neutralizing antibody (αIFN-γ) in 2 murine HLH models. In both models, ruxolitinib and αIFN-γ reduced inflammation-associated anemia, indicating that ruxolitinib operates in an IFN-γ–dependent manner to reverse this HLH manifestation. In contrast, the number and activation status of T cells and neutrophils, as well as their infiltration into tissues, were significantly reduced following treatment with ruxolitinib, but they remained unchanged or were increased following treatment with αIFN-γ. Notably, despite discontinuation of ruxolitinib, LCMV-infected Prf1−/− mice exhibited enhanced survival compared with mice in which αIFN-γ was discontinued. This protective effect could be mimicked by transient treatment with αIFN-γ and a neutrophil-depleting antibody. Thus, ruxolitinib operates through IFN-γ–dependent and -independent mechanisms to dampen HLH by targeting the deleterious effects of T cells and neutrophils, with the latter representing an unappreciated and understudied cell type that contributes to HLH pathogenesis.
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is an aggressive hyperinflammatory disorder driven by the uncontrolled activation and proliferation of CD8+ T lymphocytes and macrophages.1 HLH is defined as “primary” when it arises as a result of genetic defects affecting lymphocyte cytotoxicity and “secondary” when it arises in the absence of known genetic mutations but in the presence of an underlying trigger, such as an autoimmune condition, malignancy, or infection.1 In HLH and the related cytokine-release syndrome (CRS) occurring in patients receiving T-cell–based cancer immunotherapies, much of the associated morbidity and mortality result from the overproduction of cytokines, including interferon-γ (IFN-γ), interleukin-1 (IL-1), IL-2, IL-6, IL-12, IL-18, and tumor necrosis factor-α (TNF-α).2-5 Together, these cytokines drive immune cell activation and foster a sepsis-like syndrome that is typified by fever, cytopenias, hypotension, and tissue inflammation.
There are ongoing efforts to determine which cytokines are central to HLH development. These studies have relied heavily on the use of murine models, including lymphocytic choriomeningitis virus (LCMV) infection of perforin-deficient (Prf1−/−) mice6-8 (a model of primary HLH) or administration of the TLR9 agonist CpG, with or without IL-10 receptor blockade, to wild-type (WT) C57BL/6 mice (a model of secondary HLH).9,10 These models have revealed that neutralization of IFN-γ or blockade of IL-33 (which functions to induce or amplify IFN-γ production) lessen anemia and prolong survival.8,11-16 Together, these studies have identified IFN-γ as an essential mediator of HLH and spurred investigation of emapalumab, an IFN-γ–blocking antibody, as a treatment of children and adults with HLH (NCT03312751; NCT01818492).
Because IFN-γ and many of the cytokines elevated in HLH share overlapping signaling pathways involving the JAKs,17 our laboratory has pursued an alternative treatment approach that centers around use of the oral JAK inhibitor ruxolitinib. Toward this end, we and other investigators previously reported that ruxolitinib significantly lessens inflammation in mouse models of primary and secondary HLH.18,19 Additionally, recent case reports reveal promising results for the use of ruxolitinib as a treatment for patients with relapsed or refractory disease.20-23
Ruxolitinib inhibits JAK1 and JAK2, which function downstream of several cytokines, in addition to IFN-γ.24,25 As a result, it remained unknown whether the beneficial effects of ruxolitinib resulted simply from its targeting of IFN-γ signaling or instead from the targeting of other cytokine signaling pathways. To gain further insights, in this study we compared the therapeutic effects of ruxolitinib with those obtained using an IFN-γ–blocking antibody (αIFN-γ) in mouse models of primary and secondary HLH. We confirm that IFN-γ is the main driver of HLH-associated anemia but find that cytokines distinct from IFN-γ promote the other signs and symptoms of hyperinflammation. Supporting this observation, ruxolitinib and αIFN-γ improved hemoglobin levels and red blood cell count; however, only ruxolitinib significantly reduced the activation of, and tissue infiltration by, T cells and neutrophils. Furthermore, in the model of primary HLH, short-term treatment with ruxolitinib, but not αIFN-γ, resulted in long-lasting benefits, as demonstrated by decreased serum cytokine levels and enhanced survival, despite the discontinuation of treatment. This survival advantage could not be recapitulated by simultaneously targeting IFN-γ and individual cytokines, such as TNF-α or IL-6. Instead, short-term neutralization of IFN-γ, along with depletion of neutrophils, resulted in enhanced survival similar to that observed following transient treatment with ruxolitinib. Overall, these findings provide new insights into the mechanisms of action of ruxolitinib in HLH and reveal a hitherto unrecognized role for neutrophils in the pathogenesis of this disease. These studies further support the incorporation of ruxolitinib into future clinical trials as a rational and potentially more effective treatment for HLH and related hyperinflammatory syndromes.
Methods
Mice
Perforin-deficient (C57BL/6 Prf1tm1Sdz/J) and WT (C57BL/6-J) mice were purchased from The Jackson Laboratory. Animals were matched for age and sex and used at 6 to 12 weeks of age. Mice were housed in specific pathogen–free facilities at St. Jude Children’s Research Hospital. Experiments were conducted under the approval of the Institutional Animal Care and Use Committee.
Secondary HLH model
WT mice were injected intraperitoneally with CpG 1826 (50 μg per mouse; Integrated DNA Technologies) and 0.2 mg of an IL-10 receptor–blocking antibody (αIL-10R; clone 1B1.3A; Bio X Cell) on days 0, 2, 4, and 7.9,10 Mice were treated with 90 mg/kg ruxolitinib (from Ross Levine, Memorial Sloan Kettering Cancer Center, New York, NY), orally twice a day, as described,19 or with αIFN-γ (XMG1.2; Bio X Cell), 0.5 mg per mouse intraperitoneally on days 4 and 7 after the first CpG/αIL-10R injection.
Primary HLH model
LCMV Armstrong was provided by John Wherry (University of Pennsylvania, Philadelphia, PA). Prf1−/− mice were infected with 2 × 105 PFU LCMV via intraperitoneal injection. Mice were treated with ruxolitinib or αIFN-γ, as described above. In experiments in which cytokines or cells were depleted, the following antibodies (Bio X Cell) were administered intraperitoneally: αIFN-γ on days 4 and 7 (XMG1.2; 0.5 mg per mouse), αTNF-α on days 4, 6, and 8 (XT3.11; 1 mg per mouse), αIL-6 on days 4, 6, and 8 (MP5-20F3; 0.2 mg per mouse), and αLy6G on days 4, 6, and 8 (1A8; 0.5 mg per mouse).
Complete blood counts and serum cytokines
Heparinized blood samples were analyzed on a Forecyte multi-species hematology system (Oxford Science). Serum cytokines were measured using a MILLIPLEX MAP Mouse Cytokine/Chemokine Magnetic Bead Panel (EMD Millipore) or Quantikine ELISA (CXCL9; R&D Systems). Results were collected and analyzed with a Bio-Plex 200 System (Bio-Rad) and xPONENT software.
Histology and immunohistochemical analyses
Tissues were fixed with 10% neutral buffered formalin and embedded in paraffin. Deparaffinized sections were stained with hematoxylin and eosin (Richard-Allan Scientific) or labeled with antibodies against CD3-ε (M20; Santa Cruz Biotechnology) or Ly6B.2 (7/4; Novus Biologicals), detected with biotinylated antibodies and horseradish peroxidase–labeled streptavidin (Thermo Fisher Scientific), and counterstained with hematoxylin (Thermo Fischer Scientific). Images were acquired on a ScanScope XT scanner and digitized to scalable images up to a 20× objective (Leica Biosystems). The number of, and area encompassed by, inflammatory foci were determined using FIJI image analysis, in a blinded fashion.26
Flow cytometry and cell sorting
Splenocytes and intrahepatic leukocytes were stained with fluorescence-activated cell sorting buffer (phosphate-buffered saline [PBS], 1% fetal bovine saline, 0.05% sodium azide) for 30 minutes at 4°C using fluorescently labeled antibodies: TCRβ (H57-597), F4/80 (BM8.1), NK1.1 (PK136), Ly6C (HK1.4), CD11c (N418), CD11b (M1/70), CD8 (53-6.7), CD19-ef450 (1D3), Ly6G-BV650 (1A8), CD4 (GK1.5), CD44 (IM7), CD62L (MEL-14), TREM-1 (174031), PD-1 (29F.1A12), and Tim-3 (RMT3-23) (from eBioscience, BioLegend, Tonbo Biosciences, and R&D Systems). Staining with LCMV gp33 tetramer (National Institutes of Health Tetramer Core Facility) was performed at room temperature for 45 minutes. For intracellular cytokine staining, cells were restimulated with 0.4 μg/mL LCMV gp33-41 peptide (AnaSpec) in the presence of Brefeldin A (eBioscience) and GolgiStop (BD Biosciences) for 4 to 5 hours, per the manufacturers’ instructions. Cells were washed with FACS buffer, fixed, and permeabilized using a fixation/permeabilization solution kit (BD Biosciences) and stained with αIFN-γ (XMG1.2) and αTNF-α (MP6-XT22). Foxp3 staining kits (eBioscience or Tonbo Biosciences) were used per the manufacturers’ instructions, and cells were stained with Foxp3 antibody (FJK-15s). Cells were acquired with a BD LSR II Fortessa and analyzed using FlowJo software (v10.1r5). Neutrophils (TCRb−CD11b+Ly6CintLy6G+) were sorted with a FACSAria to a final purity >95%.
Statistical analysis
Statistical analyses were performed using GraphPad Prism. Significance was calculated using 1-way ANOVA with a post hoc Tukey’s HSD to compare multiple treatments. For survival studies, the log-rank test was performed. Outliers were removed using 1-way ANOVA with Grubb’s test.
Additional methods are available in supplemental Methods (available on the Blood Web site).
Results
Comparison of ruxolitinib and αIFN-γ in a murine model of secondary HLH
To induce secondary HLH, we used a model in which mice undergo administration of CpG oligonucleotides and αIL-10R.9,10,27 Mice receiving CpG/αIL-10R were then treated or not with ruxolitinib or αIFN-γ beginning on day 4 (Figure 1A). Animals were weighed daily and were euthanized on day 9 and analyzed for the parameters of HLH. Compared with naive mice, animals receiving CpG/αIL-10R developed weight loss (Figure 1B), thrombocytopenia (Figure 1C), and anemia (Figure 1D). This weight loss was modestly reduced following treatment with αIFN-γ, but the final weight remained 13.52% ± 1.7% less than the initial body weight (Figure 1B). In contrast, mice receiving ruxolitinib initially lost, but then regained, weight; by day 9, they weighed only 5.5% ± 1.4% less than their initial body weight (Figure 1B). Neither ruxolitinib nor αIFN-γ reversed thrombocytopenia (Figure 1C); however, both treatments reduced anemia (Figure 1D). The similar effects on red blood cell count and hemoglobin levels suggest that CpG/αIL-10R–induced anemia depends on IFN-γ, which is effectively being targeted by both treatments. In contrast, the disparate effects on weight loss and absence of improvement in thrombocytopenia suggest that these manifestations are driven by other cytokines.

Ruxolitinib (Ruxo) targets inflammation in a murine model of secondary HLH via IFN-γ–dependent and -independent mechanisms. (A) WT C57BL/6 mice were injected with PBS (Naive) or with CpG and αIL-10R, as shown. Injected mice were left untreated (UnRx) or were treated with αIFN-γ or Ruxo on days 4 to 8 after the first CpG and αIL-10R injection. On day 9, mice were euthanized and analyzed. (B) Change in body weight (as a percentage of the initial body weight) during the course of the experiment. Body weight percentage was calculated as (actual body weight/initial body weight) × 100. Peripheral blood samples were analyzed for the number of platelets (PLT) (C) and the numbers red blood cells and for the levels of hemoglobin (Hb) (D). (E) Levels of serum cytokines were determined using Luminex. (F) Splenomegaly was assessed as a percentage of body weight and calculated as (spleen weight/actual body weight) × 100. (G) Hepatomegaly was assessed as a percentage of body weight and was calculated as: (liver weight/actual body weight) × 100. Each data point represents 1 mouse, and data were collected from 3 independent experiments. Outliers were excluded using Grubb’s test. Data shown are the mean values ± standard deviation. The total number of mice per group was n = 12 each (Naive and UnRx), n = 14 (αIFN-γ), and n = 13 (Ruxo). *P < .05, **P < .01, ***P < .001, ****P < .0001.
Ruxolitinib (Ruxo) targets inflammation in a murine model of secondary HLH via IFN-γ–dependent and -independent mechanisms. (A) WT C57BL/6 mice were injected with PBS (Naive) or with CpG and αIL-10R, as shown. Injected mice were left untreated (UnRx) or were treated with αIFN-γ or Ruxo on days 4 to 8 after the first CpG and αIL-10R injection. On day 9, mice were euthanized and analyzed. (B) Change in body weight (as a percentage of the initial body weight) during the course of the experiment. Body weight percentage was calculated as (actual body weight/initial body weight) × 100. Peripheral blood samples were analyzed for the number of platelets (PLT) (C) and the numbers red blood cells and for the levels of hemoglobin (Hb) (D). (E) Levels of serum cytokines were determined using Luminex. (F) Splenomegaly was assessed as a percentage of body weight and calculated as (spleen weight/actual body weight) × 100. (G) Hepatomegaly was assessed as a percentage of body weight and was calculated as: (liver weight/actual body weight) × 100. Each data point represents 1 mouse, and data were collected from 3 independent experiments. Outliers were excluded using Grubb’s test. Data shown are the mean values ± standard deviation. The total number of mice per group was n = 12 each (Naive and UnRx), n = 14 (αIFN-γ), and n = 13 (Ruxo). *P < .05, **P < .01, ***P < .001, ****P < .0001.
Supporting this possibility, mice receiving CpG/αIL-10R exhibited elevated serum levels of many proinflammatory cytokines, including IFN-γ and its downstream effectors CXCL9 and CXCL10, as well as IL-6, IL-12, TNF-α, granulocyte macrophage colony-stimulating factor (GM-CSF), monocyte chemoattractant protein-1, and macrophage inflammatory protein-1α (Figure 1E; supplemental Figure 1). Because several of these cytokines signal through JAK1 (eg, IFN-γ, IL-6) or JAK2 (eg, IL-12, GM-CSF) or are induced by cytokines operating via the JAK-STAT pathway (eg, IFN-α, IFN-β, IL-15),28,29 we hypothesized that they and their impacts on the immune system (eg, splenic or hepatic enlargement) would be lessened by treatment with ruxolitinib. Indeed, ruxolitinib was superior to αIFN-γ in lowering the levels of cytokines and reducing organomegaly (Figure 1F-G).
Comparison of ruxolitinib and IFN-γ neutralization in a murine model of primary HLH
To examine the effects of ruxolitinib and αIFN-γ in a model of primary HLH, Prf1−/− mice were infected with LCMV and were left untreated or were treated with either agent starting on day 4 postinfection (Figure 2A). On day 9, mice were euthanized and evaluated for HLH. Unlike naive mice, LCMV-infected animals developed weight loss (Figure 2B), cytopenias (Figure 2C-D), organomegaly (Figure 2F-G), and increased serum cytokine levels (Figure 2E; supplemental Figure 1). Among these cytokines, IFN-γ exhibited the highest levels, in line with its known role in driving LCMV-induced inflammation. Once again, ruxolitinib and αIFN-γ lessened anemia (Figure 2D); however, only ruxolitinib significantly diminished serum cytokine levels (Figure 2E) and reduced splenic and hepatic enlargement (Figure 2F-G). To ensure sufficient inhibition of IFN-γ activity, studies were repeated using twice the dose of αIFN-γ (1 mg instead of 0.5 mg), yet the same results were obtained (supplemental Figure 2A-D). Nevertheless, both doses completely normalized STAT1 phosphorylation in peripheral blood monocytes from LCMV-infected mice. In addition, incubation of RAW264.7 macrophages with sera from mice treated with either dose of αIFN-γ failed to induce STAT1 phosphorylation or expression of IFN-γ–induced GTPase transcript (supplemental Figure 1F-G). Therefore, regardless of the αIFN-γ dose used, IFN-γ activity appeared fully neutralized. Together with findings from the secondary HLH model, these observations reveal that ruxolitinib operates in an IFN-γ–dependent manner to reverse inflammation-induced anemia but in an IFN-γ–independent manner to alleviate hypercytokinemia and organomegaly.
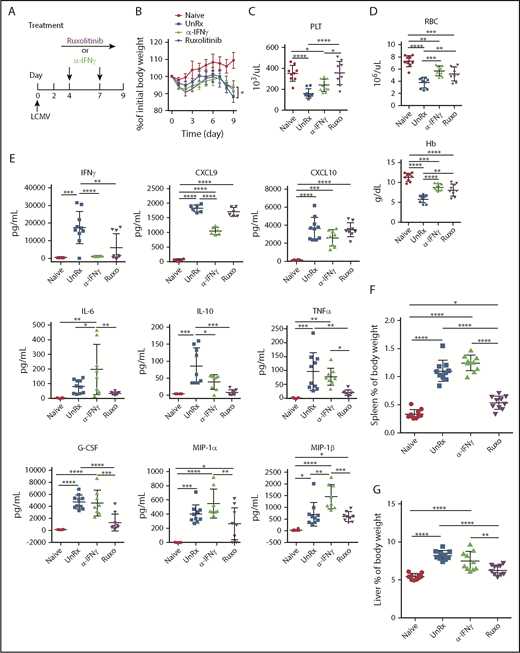
Ruxolitinib (Ruxo) targets inflammation in a murine model of primary HLH via IFN-γ–dependent and -independent mechanisms. (A) LCMV-infected Prf1−/− mice were left untreated (UnRx) or were treated with αIFN-γ or Ruxo, as shown. On day 9, mice were euthanized and analyzed. Uninfected Prf1−/− mice (Naive) were used as a control. (B) Change in body weight (as a percentage of initial body weight) during the course of the experiment. Body weight percentage was calculated as (actual body weight/ initial body weight) × 100. Peripheral blood samples were analyzed for the number of platelets (PLT) (C) and the number of red blood cells (RBC) and for the levels of hemoglobin (Hb) (D). (E) Levels of serum cytokines were determined using Luminex. (F) Splenomegaly was assessed as a percentage of body weight and calculated as (spleen weight/actual body weight) × 100. (G) Hepatomegaly was assessed as a percentage of overall body weight and was calculated as (liver weight/actual body weight) × 100. Each data point represents 1 mouse. Data were collected from 2 independent experiments and are shown are the mean values ± standard deviation. The total number of mice per group was n = 10 each for Naive, UnRx, αIFN-γ, and Ruxo. For cytokine analysis, the total number of mice per group was n = 6 (Naive), n = 10 (UnRx), n = 8 (αIFN-γ), and n = 9 (Ruxo). Outliers were excluded using Grubb’s test. *P < .05, **P < .01, ***P < .001, ****P < .0001.
Ruxolitinib (Ruxo) targets inflammation in a murine model of primary HLH via IFN-γ–dependent and -independent mechanisms. (A) LCMV-infected Prf1−/− mice were left untreated (UnRx) or were treated with αIFN-γ or Ruxo, as shown. On day 9, mice were euthanized and analyzed. Uninfected Prf1−/− mice (Naive) were used as a control. (B) Change in body weight (as a percentage of initial body weight) during the course of the experiment. Body weight percentage was calculated as (actual body weight/ initial body weight) × 100. Peripheral blood samples were analyzed for the number of platelets (PLT) (C) and the number of red blood cells (RBC) and for the levels of hemoglobin (Hb) (D). (E) Levels of serum cytokines were determined using Luminex. (F) Splenomegaly was assessed as a percentage of body weight and calculated as (spleen weight/actual body weight) × 100. (G) Hepatomegaly was assessed as a percentage of overall body weight and was calculated as (liver weight/actual body weight) × 100. Each data point represents 1 mouse. Data were collected from 2 independent experiments and are shown are the mean values ± standard deviation. The total number of mice per group was n = 10 each for Naive, UnRx, αIFN-γ, and Ruxo. For cytokine analysis, the total number of mice per group was n = 6 (Naive), n = 10 (UnRx), n = 8 (αIFN-γ), and n = 9 (Ruxo). Outliers were excluded using Grubb’s test. *P < .05, **P < .01, ***P < .001, ****P < .0001.
Ruxolitinib reduces T-cell and neutrophil accumulation in inflamed tissues
To explore the mechanisms through which ruxolitinib was influencing CpG/αIL-10R–induced inflammation and to determine their dependence upon the inhibition of IFN-γ, we quantified the frequency and absolute numbers of immune cell populations within the organs of untreated mice and mice that had received ruxolitinib or αIFN-γ (supplemental Figure 3; gating strategy). Mice administered CpG/αIL-10R exhibited a modest increase in the frequency and number of splenic CD8+ T cells (Figure 3A). Consistent with prior work showing that innate immune cell activation is central to disease development in this model,10,30 we observed a dramatic increase in the frequency and number of myeloid cells, particularly neutrophils, in the spleen (Figure 3B). These mice also had increased accumulation of T cells and neutrophils in the liver (Figure 3C-D). Compared with αIFN-γ, which had little impact on T-cell or myeloid cell accumulation, ruxolitinib significantly reduced the frequency and/or absolute number of organ-infiltrating CD8+ cells, monocytes, and neutrophils (Figure 3).

Ruxolitinib (Ruxo) reduces the expansion of T cells and neutrophils in secondary HLH. (A) Frequency (upper panels) and absolute numbers (lower panels) of splenic CD8+ T cells (left panels) and CD4+ T cells (right panels) gated on CD19−TCRβ+ cells. (B) Representative flow cytometric plots showing Ly6ChiLy6G− monocytes and Ly6G+Ly6Cint neutrophils gated on CD19−TCRβ−NK1.1−CD11c−CD11b+ cells. Summarized data are the frequency (upper right panels) and absolute numbers (lower right panels) of splenic monocytes and neutrophils. Each data point represents 1 mouse, and data were collected from 3 independent experiments. The mean ± standard deviation are shown. (C) Representative images showing hematoxylin and eosin–stained sections of liver (top row) and immunohistochemical-stained sections of liver showing CD3+ (middle row) and Neut7-4+ (bottom row) cells from mice injected with PBS (Naive) or CpG and αIL-10R that were left untreated (UnRx) or treated with αIFN-γ or Ruxo. Original magnification ×200. The total number of mice per group was n = 12 each (Naive and UnRx), n = 14 (IFN-γ) and n = 13 (Ruxo). (D) Summarized data from liver histological analyses showing the area of tissue infiltrated by immune cells (upper left panel), the number of inflammatory foci per field of view at 2× magnification (upper right panel) and the percentages of CD3+ cells (lower left panel) and Neut7-4+ cells (lower right panel) within inflammatory foci. Data were collected from 2 independent experiments (n = 4 mice per group). Samples were randomly chosen for histological analysis. *P < .05, **P < .01, ***P < .001, ****P < .0001.
Ruxolitinib (Ruxo) reduces the expansion of T cells and neutrophils in secondary HLH. (A) Frequency (upper panels) and absolute numbers (lower panels) of splenic CD8+ T cells (left panels) and CD4+ T cells (right panels) gated on CD19−TCRβ+ cells. (B) Representative flow cytometric plots showing Ly6ChiLy6G− monocytes and Ly6G+Ly6Cint neutrophils gated on CD19−TCRβ−NK1.1−CD11c−CD11b+ cells. Summarized data are the frequency (upper right panels) and absolute numbers (lower right panels) of splenic monocytes and neutrophils. Each data point represents 1 mouse, and data were collected from 3 independent experiments. The mean ± standard deviation are shown. (C) Representative images showing hematoxylin and eosin–stained sections of liver (top row) and immunohistochemical-stained sections of liver showing CD3+ (middle row) and Neut7-4+ (bottom row) cells from mice injected with PBS (Naive) or CpG and αIL-10R that were left untreated (UnRx) or treated with αIFN-γ or Ruxo. Original magnification ×200. The total number of mice per group was n = 12 each (Naive and UnRx), n = 14 (IFN-γ) and n = 13 (Ruxo). (D) Summarized data from liver histological analyses showing the area of tissue infiltrated by immune cells (upper left panel), the number of inflammatory foci per field of view at 2× magnification (upper right panel) and the percentages of CD3+ cells (lower left panel) and Neut7-4+ cells (lower right panel) within inflammatory foci. Data were collected from 2 independent experiments (n = 4 mice per group). Samples were randomly chosen for histological analysis. *P < .05, **P < .01, ***P < .001, ****P < .0001.
Much of the immunopathology associated with LCMV infection of Prf1−/− mice is believed to result from the unchecked activation of CD8+ T cells, which accumulate in infected organs and cause significant tissue damage.8,16 However, studies using WT mice reveal that neutrophils are also recruited to sites of LCMV infection.31-34 Based on this information, we next sought to investigate the contribution of T cells and neutrophils during primary HLH and the effects of ruxolitinib treatment upon them. Therefore, we quantified these lineages in the organs of LCMV-infected Prf1−/− mice that had been treated or not with ruxolitinib or αIFN-γ. Consistent with prior reports,8,19 we observed a marked increase in the frequency and absolute number of CD8+ T cells in the spleens (Figure 4A-B) and livers (Figure 4C-D) of LCMV-infected animals. Extending prior observations, we also observed a significant increase in the percentage and number of neutrophils and monocytes in the spleen (Figure 4B) and an increased percentage of neutrophils within inflammatory foci in the liver (Figure 4C-D). Compared with untreated animals, ruxolitinib significantly reduced the numbers of splenic CD8+ T cells (Figure 4A) and neutrophils (Figure 4B) and lowered their frequency within intrahepatic inflammatory foci (Figure 4C-D). In contrast, αIFN-γ did not significantly diminish these cell populations in either of the organs examined.
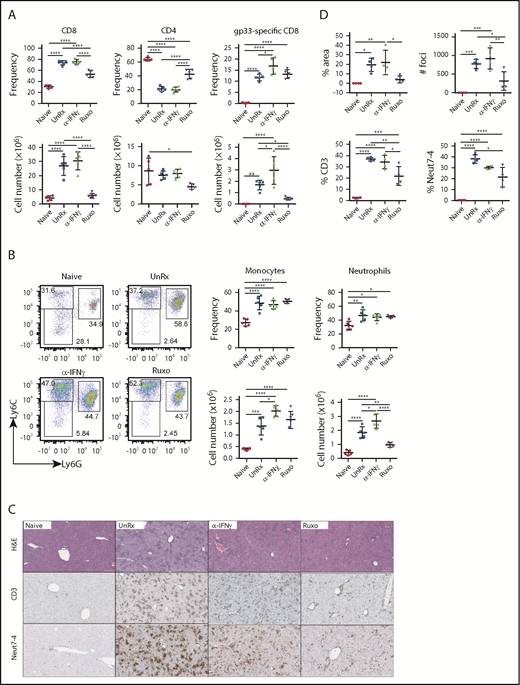
Ruxolitinib (Ruxo) reduces the expansion of T cells and neutrophils in primary HLH. (A) Summarized data of the frequency and absolute numbers of splenic CD8+ T cells (left panels), CD4+ T cells gated on TCRb+CD19− live cells (middle panels), and LCMV-specific (Db gp33) CD8+ T cells gated on CD8+ T cells (right panels). (B) Frequency of splenic monocytes (Ly6ChiLy6G−) and neutrophils (Ly6CintLy6G+) of CD11b+CD11c− cells. Data (mean ± standard deviation) are representative of 2 independent experiments. n = 5 mice per group. (C) Representative hematoxylin and eosin–stained liver sections (top row) and immunohistochemical staining of CD3+ (middle row) and Neut7-4+ (lower row) cells from naive Prf1−/− mice or mice infected with LCMV that were left untreated (UnRx) or treated with αIFN-γ or Ruxo. Original magnification ×200. (D) Data were quantitated and plotted as percentage area of inflammation (upper left panel), number of inflammatory foci per field of view at 2× magnification (upper right panel) and the percentages of Neut7-4+ cells (lower left panel) and CD3+ cells (lower right panel). Data were collected from 2 independent experiments (n = 4 mice per group). Samples were randomly chosen for histological analysis. *P < .05, **P < .01, ***P < .001, ****P < .0001.
Ruxolitinib (Ruxo) reduces the expansion of T cells and neutrophils in primary HLH. (A) Summarized data of the frequency and absolute numbers of splenic CD8+ T cells (left panels), CD4+ T cells gated on TCRb+CD19− live cells (middle panels), and LCMV-specific (Db gp33) CD8+ T cells gated on CD8+ T cells (right panels). (B) Frequency of splenic monocytes (Ly6ChiLy6G−) and neutrophils (Ly6CintLy6G+) of CD11b+CD11c− cells. Data (mean ± standard deviation) are representative of 2 independent experiments. n = 5 mice per group. (C) Representative hematoxylin and eosin–stained liver sections (top row) and immunohistochemical staining of CD3+ (middle row) and Neut7-4+ (lower row) cells from naive Prf1−/− mice or mice infected with LCMV that were left untreated (UnRx) or treated with αIFN-γ or Ruxo. Original magnification ×200. (D) Data were quantitated and plotted as percentage area of inflammation (upper left panel), number of inflammatory foci per field of view at 2× magnification (upper right panel) and the percentages of Neut7-4+ cells (lower left panel) and CD3+ cells (lower right panel). Data were collected from 2 independent experiments (n = 4 mice per group). Samples were randomly chosen for histological analysis. *P < .05, **P < .01, ***P < .001, ****P < .0001.
Ruxolitinib reduces T-cell and neutrophil activation in LCMV-infected Prf1−/− mice
Having shown that ruxolitinib lessens the expansion of T cells and neutrophils, we next sought to examine whether it also dampens their activation. To assess T-cell activation, splenocytes were harvested on day 9 from naive mice, LCMV-infected mice, or LCMV-infected mice that had been treated with αIFN-γ or ruxolitinib. Splenocytes were incubated with an MHC class I–restricted LCMV peptide and, 5 hours later, CD8+ T cells were examined for intracellular cytokine production. In contrast to the CD8+ T cells from naive mice, a large proportion of cells from LCMV-infected animals produced TNF-α and/or IFN-γ (Figure 5A). In comparison, many fewer cells from ruxolitinib-treated animals did so (Figure 5A). Notably, αIFN-γ exerted the opposite effect, with significantly more CD8+ T cells producing 1 or both cytokines (Figure 5A). Furthermore, the CD8+ T cells from αIFN-γ−treated animals produced much higher levels of cytokines on a per-cell basis than did the T cells from ruxolitinib-treated animals (Figure 5B-C).
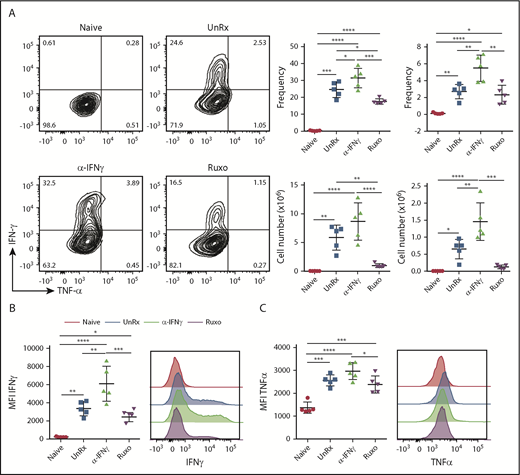
Ruxolitinib (Ruxo) reduces CD8+ T-cell cytokine production in primary HLH. (A) Representative flow cytometric plots of intracellular IFN-γ and TNF-α produced by splenic CD8+ T cells after in vitro stimulation with LCMV-restricted gp33 peptide. Depicted on the right are summarized frequency (upper panels) and absolute numbers (lower panels) of TNF-α−IFN-γ+ and TNF-α+IFN-γ+ CD8+ T cells. (B) Mean fluorescence intensity (MFI) of IFN-γ in CD8+ T cells (left panel) and representative graphs (right panel). (C) MFI of TNF-α in CD8+ T cells (left panel) and representative graphs (right panel). Each data point represents 1 mouse. Data (mean ± standard deviation) are representative of 2 independent experiments (n = 5 mice per group). *P < .05, **P < .01, ***P < .001, ****P < .0001.
Ruxolitinib (Ruxo) reduces CD8+ T-cell cytokine production in primary HLH. (A) Representative flow cytometric plots of intracellular IFN-γ and TNF-α produced by splenic CD8+ T cells after in vitro stimulation with LCMV-restricted gp33 peptide. Depicted on the right are summarized frequency (upper panels) and absolute numbers (lower panels) of TNF-α−IFN-γ+ and TNF-α+IFN-γ+ CD8+ T cells. (B) Mean fluorescence intensity (MFI) of IFN-γ in CD8+ T cells (left panel) and representative graphs (right panel). (C) MFI of TNF-α in CD8+ T cells (left panel) and representative graphs (right panel). Each data point represents 1 mouse. Data (mean ± standard deviation) are representative of 2 independent experiments (n = 5 mice per group). *P < .05, **P < .01, ***P < .001, ****P < .0001.
To examine neutrophil activation, splenocytes were harvested on day 9, and neutrophils were examined for expression of the activating receptor triggering receptor expressed on myeloid cells-1 (TREM-1) and for intracellular production of TNF-α. Sort-purified neutrophils were also examined for spontaneous release of cytokines after overnight culture. Following LCMV infection, neutrophils upregulated the expression of TREM-1 (Figure 6A), and a small proportion produced TNF-α (Figure 6B). Sort-purified neutrophils exhibited enhanced production of cytokines, including CXCL10, MIP-1α, MIP-1β, and IL-1β (supplemental Figure 4A-B). On the other hand, many fewer neutrophils from ruxolitinib-treated animals expressed TREM-1, TNF-α, or IL-1β (Figure 6; supplemental Figure 4B). Neutrophils from αIFN-γ–treated animals exhibited a mixed response, with more cells expressing TREM-1 and IL-1β and fewer producing TNF-α. Altogether, these data on cell expansion and activation reveal that ruxolitinib lessens CpG/αIL-10R– and LCMV-induced inflammation via its potent effects on T cells, as well as myeloid cells, especially neutrophils. Compared with αIFN-γ, the more pronounced effects of ruxolitinib on cell number and function strongly suggest that its mechanisms of action extend beyond the mere inhibition of IFN-γ signaling.
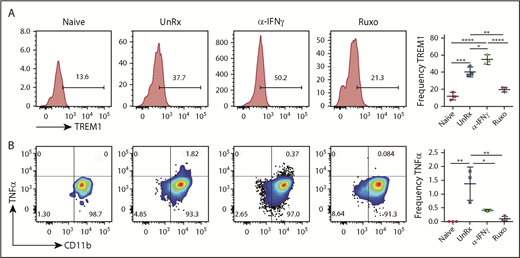
Ruxolitinib (Ruxo) reduces neutrophil activation in primary HLH. (A) Representative plots showing surface expression of TREM-1 on neutrophils. Graph depicts the frequency of splenic TREM-1+ neutrophils (right panel). (B) Representative plots showing intracellular TNF-α staining in splenic neutrophils gated on TCRb−CD11b+Ly6CintLy6G+ cells from naive or LCMV-infected Prf1−/− mice that were treated or not with αIFN-γ or Ruxo. Graph depicts the frequency of TNF-α+ neutrophils (right panel). Data (mean ± standard deviation) are representative of 2 independent experiments (n = 3 mice per group). *P < .05, **P < .01, ***P < .001, ****P < .0001.
Ruxolitinib (Ruxo) reduces neutrophil activation in primary HLH. (A) Representative plots showing surface expression of TREM-1 on neutrophils. Graph depicts the frequency of splenic TREM-1+ neutrophils (right panel). (B) Representative plots showing intracellular TNF-α staining in splenic neutrophils gated on TCRb−CD11b+Ly6CintLy6G+ cells from naive or LCMV-infected Prf1−/− mice that were treated or not with αIFN-γ or Ruxo. Graph depicts the frequency of TNF-α+ neutrophils (right panel). Data (mean ± standard deviation) are representative of 2 independent experiments (n = 3 mice per group). *P < .05, **P < .01, ***P < .001, ****P < .0001.
Transient treatment with ruxolitinib, but not αIFN-γ, enhances long-term survival
A prior study has shown that discontinuous treatment with ruxolitinib (eg, 14 days then stop) enhances survival of LCMV-infected Prf1−/− mice.18 To determine the extent to which this relies on the dampening of IFN-γ signaling, Prf1−/− mice were infected or not with LCMV and then treated or not with ruxolitinib or αIFN-γ. On day 9, ruxolitinib and αIFN-γ were discontinued, and animals were monitored until day 35 postinfection (Figure 7A). All untreated LCMV-infected animals succumbed to disease (Figure 7A). However, upon stopping ruxolitinib, all 14 mice (100%) survived (Figure 7A; Ruxo). This differed from mice treated with αIFN-γ; only 5 of 13 (38.5%) animals lived to the end point of the experiment. Different viral loads did not explain this discrepancy in outcome, because viral titers were similarly elevated in mice from both treatment cohorts (data not shown). Thus, early short-term treatment with ruxolitinib results in long-lasting clinical effects that are more pronounced in mice receiving ruxolitinib vs those receiving αIFN-γ.
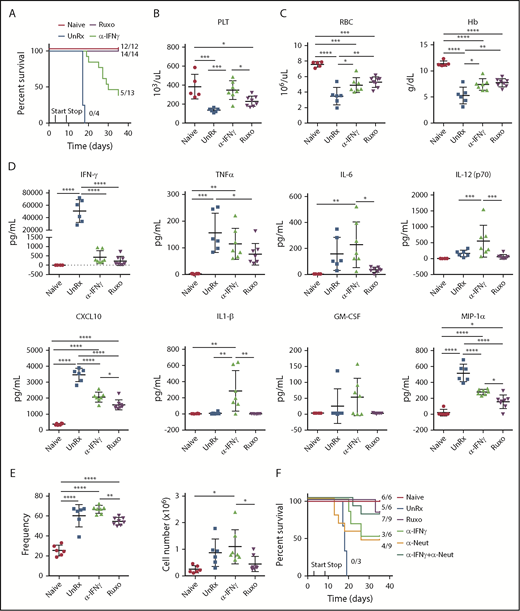
Short-term treatment with ruxolitinib (Ruxo) promotes long-term survival of LCMV-infected Prf1−/−mice. (A) Naive and LCMV-infected Prf1−/− mice were left untreated (UnRx) or were treated with αIFN-γ on days 4 and 7 postinfection or with Ruxo on days 4 to 8 postinfection. αIFN-γ and Ruxo were discontinued on day 9, and survival was monitored until day 35. Data are combined from 3 independent experiments. P < .0001, log-rank test. (B) Mice were treated as in (A) and euthanized on day 20. For comparison, untreated LCMV-infected mice were euthanized and analyzed on day 9 (UnRx). Blood was analyzed for platelet count (PLT) (B) and for red blood cell count (RBC) and hemoglobin (Hb) (C). (D) Levels of serum cytokines. (E) Frequency (left panel) and total numbers (right panel) of splenic neutrophils on day 20 postinfection. Each data point represents 1 mouse. Data (mean ± standard deviation) are combined from 2 independent experiments. The total number of mice per group was n = 6 each (Naive and UnRx), n = 7 (αIFN-γ) and n = 8 (Ruxo). (F) Percentage survival of Naive and LCMV-infected Prf1−/− mice left untreated (UnRx) or treated with αIFN-γ, Ruxo, neutrophil-depleting antibody (αNeut), or a combination of αNeut and αIFN-γ from days 4 to 8 postinfection, followed by treatment discontinuation. Survival was followed to day 35. Data (mean ± standard deviation) are combined from 2 independent experiments. P < .0004, log-rank test. Total number of mice examined was n = 6 each (Naive, αIFN-γ, and Ruxo), n = 3 (UnRx), and n = 9 each (αNeut and αNeut+αIFN-γ). *P < .05, **P < .01, ***P < .001, **** P < .0001.
Short-term treatment with ruxolitinib (Ruxo) promotes long-term survival of LCMV-infected Prf1−/−mice. (A) Naive and LCMV-infected Prf1−/− mice were left untreated (UnRx) or were treated with αIFN-γ on days 4 and 7 postinfection or with Ruxo on days 4 to 8 postinfection. αIFN-γ and Ruxo were discontinued on day 9, and survival was monitored until day 35. Data are combined from 3 independent experiments. P < .0001, log-rank test. (B) Mice were treated as in (A) and euthanized on day 20. For comparison, untreated LCMV-infected mice were euthanized and analyzed on day 9 (UnRx). Blood was analyzed for platelet count (PLT) (B) and for red blood cell count (RBC) and hemoglobin (Hb) (C). (D) Levels of serum cytokines. (E) Frequency (left panel) and total numbers (right panel) of splenic neutrophils on day 20 postinfection. Each data point represents 1 mouse. Data (mean ± standard deviation) are combined from 2 independent experiments. The total number of mice per group was n = 6 each (Naive and UnRx), n = 7 (αIFN-γ) and n = 8 (Ruxo). (F) Percentage survival of Naive and LCMV-infected Prf1−/− mice left untreated (UnRx) or treated with αIFN-γ, Ruxo, neutrophil-depleting antibody (αNeut), or a combination of αNeut and αIFN-γ from days 4 to 8 postinfection, followed by treatment discontinuation. Survival was followed to day 35. Data (mean ± standard deviation) are combined from 2 independent experiments. P < .0004, log-rank test. Total number of mice examined was n = 6 each (Naive, αIFN-γ, and Ruxo), n = 3 (UnRx), and n = 9 each (αNeut and αNeut+αIFN-γ). *P < .05, **P < .01, ***P < .001, **** P < .0001.
To decipher the mechanisms underlying these findings, we examined immunologic parameters on day 20 postinfection, the point at which the survival curves for LCMV-infected mice transiently exposed to ruxolitinib or αIFN-γ began to diverge (Figure 7A). These parameters were compared with those of naive mice, as well as LCMV-infected animals that had received no treatment (this cohort was assessed on day 9 prior to becoming moribund). On day 20, mice transiently exposed to ruxolitinib or αIFN-γ exhibited similar improvements in anemia (Figure 7B-C) and comparable reductions in serum IFN-γ and CXCL10 (Figure 7D). Furthermore, both cohorts of mice had similar numbers of splenic T cells, and these T cells displayed an exhausted phenotype (supplemental Figure 5A-C). Ruxolitinib increases the number of T regulatory cells in a murine model of graft-versus-host-disease.35 Therefore, we examined whether this might underlie the improved survival in mice transiently exposed to ruxolitinib; however, we found that the numbers of T regulatory cells and the ratios of T regulatory cells/effector CD8+ T cells were similar in mice transiently exposed to ruxolitinib or αIFN-γ (supplemental Figure 5D). These results suggest that the protective properties of ruxolitinib are not dependent upon the reversal of anemia or lessening of IFN-γ, nor do they appear to be due to effects on T-cell number and/or function.
Compared with transient exposure to αIFN-γ, transient exposure to ruxolitinib significantly lowered the levels of innate-type cytokines, such as TNF-α, IL-6, IL-12, CXCL10, IL-1β, GM-CSF, and MIP-1α (Figure 7D). Because TNF-α and IL-6 blockade are used to treat secondary HLH and CRS, we examined whether the transient blockade of either of these cytokines, in addition to the transient blockade of IFN-γ, might improve survival of LCMV-infected Prf1−/− mice. However, this did not confer any added benefit over transient blockade of αIFN-γ (supplemental Figure 6).
In our earlier experiments, we observed a large influx of activated neutrophils into the organs of HLH-affected animals, which was greatly attenuated following ruxolitinib treatment. In mice transiently exposed to ruxolitinib and followed to day 20, we observed reduced frequency and numbers of neutrophils in the spleen (Figure 7E), yet these results were less apparent in αIFN-γ–treated animals. Thus, to examine whether the superior survival of LCMV-infected mice transiently exposed to ruxolitinib might be due to its suppressive effect on neutrophils, we administered a neutrophil-depleting antibody (supplemental Figure 4C) alone or with αIFN-γ on days 4 to 8 following LCMV infection. Both antibodies were then discontinued, and animals were monitored until day 35 for survival. Only 50% of mice transiently treated with αIFN-γ and 44% of mice treated with neutrophil-depleting antibody lived to day 35. On the other hand, 78% of mice treated with αIFN-γ and the neutrophil-depleting antibody survived (Figure 7F), a value comparable to the 83% survival of mice transiently treated with ruxolitinib in these experiments. Overall, these data support a central role for neutrophils in driving LCMV-induced HLH and demonstrate the unique ability of ruxolitinib to target this cell population and, thus, mitigate hyperinflammation.
Discussion
HLH and related CRS remain significant clinical challenges, with high morbidity and mortality, demanding more effective treatments. The mechanisms underlying these disorders are not well understood; however, 1 of the key factors driving inflammation is the excessive production of IFN-γ. Based on this observation, prior preclinical studies have shown that targeting IFN-γ through the use of neutralizing antibodies11,15 or, more recently, through the JAK1/2 inhibitor ruxolitinib18,19 significantly lessens inflammation. Nevertheless, because ruxolitinib targets signaling downstream of numerous proinflammatory cytokines, it remained unknown whether the beneficial influences of this drug were due solely to the inhibition of IFN-γ or to the targeting of other cytokines. In this study, we demonstrate that ruxolitinib, when given at a dose that blocks IFN-γ activity in a manner comparable to an IFN-γ neutralizing antibody, is significantly more effective in dampening inflammation. We also show that it functions through mechanisms that extend beyond the mere inhibition of IFN-γ signaling. Notably, compared with αIFN-γ, ruxolitinib suppressed innate, as well as adaptive, immune cell activation and resulted in long-lasting immunomodulatory effects.
This study brings to light several novel observations. First, it reveals that targeting JAK1/2-dependent cytokines represents an effective means to treat HLH. By blocking cytokines that stimulate the innate (IFN-α, IFN-β, IL-6, granulocyte colony-stimulating factor [G-CSF], GM-CSF) and adaptive (IL-2, IL-12, IFN-γ) immune systems, JAK inhibitors, such as ruxolitinib, provide an attractive therapy for these and other cytokine-driven disorders. Accordingly, there is growing evidence that treatment with JAK inhibitors is beneficial in patients with myeloproliferative disorders,36,37 rheumatoid arthritis,38,39 ulcerative colitis,40 graft-versus-host disease,35 or autoinflammatory interferonopathies.41 Our promising results support further investigations of JAK inhibition in humans with HLH and CRS.
Second, this study provides insights into the pathogenesis of various murine HLH models and the mechanisms of action of ruxolitinib and αIFN-γ upon the disease. For example, despite treatment with ruxolitinib and αIFN-γ at doses that block IFN-γ activity to comparable degrees, ruxolitinib lowers serum IFN-γ levels in the primary and secondary HLH models, whereas αIFN-γ lowers IFN-γ levels only in the primary model. This difference might reflect the distinct biologic mechanisms underlying these 2 HLH models. Specifically, development of disease in the primary model is strongly dependent upon IFN-γ.8 In contrast, development of disease in the secondary model is less dependent upon IFN-γ, because hyperinflammation can readily be provoked in IFN-γ–knockout mice.9 Therefore, although the neutralization of IFN-γ might be sufficient to lessen disease (and, thus, lower serum IFN-γ levels) in the primary model, it may not be sufficient to do so in the secondary model. Although IFN-γ remains elevated in the secondary model, we believe that its activity has been effectively neutralized by αIFN-γ. Finally, we propose that the modestly elevated IFN-γ levels remain detectable in the secondary model, because αIFN-γ recognizes an epitope that is distinct from the 1 recognized by the antibody used in the IFN-γ enzyme-linked immunosorbent assay (ie, the assay is detecting circulating antibody–IFN-γ complexes that are functionally inactive).
In both HLH models, anemia was ameliorated following treatment with ruxolitinib or αIFN-γ. Anemia results from prolonged exposure to IFN-γ; studies in mice have shown that IFN-γ acts directly on macrophages to stimulate the uptake of red blood cells via endocytosis.15 IFN-γ also inhibits the proliferation of immature bone marrow and splenic erythroid progenitors.9,42 The JAK-dependent cytokine IL-12 is a strong inducer of IFN-γ production, and neutralization of IL-12 ameliorates anemia similar to αIFN-γ.9 Thus, the alleviation of anemia in ruxolitinib-treated mice likely results from the inhibition of signaling downstream of IL-12 and/or IFN-γ.
Unlike anemia, T-cell and myeloid cell accumulation and activation, as well as the serum levels of several proinflammatory cytokines, were significantly reduced with ruxolitinib but were unaffected or even augmented in mice treated with αIFN-γ. It is perhaps not surprising that there is less activity of αIFN-γ on systemic inflammation, because many cytokines, in addition to IFN-γ, are elevated in patients with HLH.4 In fact, the severity of HLH often correlates with the levels of specific cytokines, such as IL-2, IL-6, IL-8, IL-12, MCP-1, and TNF-α, in the blood.2,3,43,44 Thus, by dampening signaling mediated by many of these cytokines, ruxolitinib likely exerts a more pronounced impact.
Other than IFN-γ, it has been difficult to determine which of the cytokines targeted by ruxolitinib are responsible for its beneficial influence on hyperinflammation, particularly after treatment discontinuation. In both murine models, ruxolitinib significantly diminished the levels of TNF-α and IL-6, cytokines that serve as alternative therapeutic targets for patients with HLH.5,45-47 IFN-γ induces TNF-α production by myeloid cells,48,49 so the reduction in serum TNF-α levels might be partially explained by ruxolitinib-mediated inhibition of IFN-γ signaling. Ruxolitinib also lowered the serum levels of G-CSF, a cytokine elevated in the primary HLH model. Because high G-CSF levels are associated with neutrophilia and thrombocytopenia,50 it is possible that the reduction in G-CSF levels improved these disease parameters.
Curiously, we observed that short-term treatment of Prf1−/− mice with ruxolitinib during the course of LCMV infection curtailed inflammatory responses and enhanced survival, despite the discontinuation of treatment. This finding is consistent with an earlier report in which treatment with ruxolitinib for 14 days, followed by stopping the drug, allowed survival of infected mice for an additional week (the end point of these studies).18 In our model, the superior survival of mice transiently exposed to ruxolitinib was not due to the preferential induction of T regulatory cells, a phenomenon previously reported for ruxolitinib,35 nor was it due to the emergence of T-cell exhaustion. Instead, transient exposure to ruxolitinib significantly decreased the serum levels of innate-type cytokines and the frequency and number of neutrophils, suggesting that continued neutrophil accumulation or activity underlies the poorer survival of mice transiently exposed to αIFN-γ. Consistent with this notion, concomitant neutrophil depletion and transient neutralization of IFN-γ enhanced survival similar to what was observed following transient exposure to ruxolitinib.
We find that neutrophils exhibit an activated phenotype and are recruited to inflamed organs, regardless of the HLH model. Neutrophils play a role during LCMV infection of WT mice,34 and prior studies have revealed that activated neutrophils mediate vascular damage, leading to lethality following intracranial LCMV infection.32 However, little attention has been given to neutrophils in the pathogenesis of human HLH, because neutropenia is a common manifestation of the disease.51 To explore the role of neutrophils in human HLH, we analyzed tissue samples from several cases of confirmed HLH or HLH-like illnesses, many of which occurred in the setting of infection. Although neutrophils were observed in a few of the samples, overall, their numbers were lower than in the murine model (supplemental Figure 7). Despite these findings, the neutrophil response may be robust early in the course of HLH, before patients come to medical attention. In agreement with this possibility, gene expression analyses of peripheral blood mononuclear cells from HLH patients reveal high levels of expression of IL-8 and TREM-1,52 both of which are important mediators of neutrophil recruitment and activation.31,53,54
In summary, through these studies we demonstrate that ruxolitinib effectively dampens inflammation in HLH and enhances outcomes via mechanisms that are only partially dependent upon the inhibition of IFN-γ signaling. Furthermore, we identify a previously unappreciated and central role for neutrophils in the pathogenesis of HLH, a cell lineage that is effectively targeted by ruxolitinib. Altogether, these studies support further investigations of ruxolitinib as a rational and potentially more effective treatment of children and adults with HLH and related CRS.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the St. Jude Flow Cytometry Core Facility for cell sorting, Peter Schreiner in the Department of Computational Biology for assistance with statistical analyses of the data, and the Pathology Department at University of Pittsburgh Medical Center Children’s Hospital of Pittsburgh for technical assistance.
This work was supported by grants from the National Institutes of Health, National Institute of Allergy and Infectious Diseases (K.E.N.), Histiocytosis Association of America (K.E.N.), and American Lebanese Syrian Associated Charities.
Authorship
Contribution: S.A. and K.C.V. designed and performed the experiments and wrote the manuscript; P.E.T. and R.B. provided technical assistance and carried out experiments; H.T. completed histological analyses of murine tissues; J.P. completed histological analyses of human spleens; and K.E.N. oversaw the project, interpreted data, and edited the manuscript.
Conflict-of-interest disclosure: K.E.N. receives research funding from Incyte Pharmaceuticals and Alpine Biosciences. The remaining authors declare no competing financial interests
Correspondence: Kim E. Nichols, Division of Cancer Predisposition, St. Jude Children’s Research Hospital, 262 Danny Thomas Place, Memphis, TN 38105; e-mail: kim.nichols@stjude.org.
