

Key Points
Clonal expansion in ATLL patients who progressed after nivolumab exposed ATLL cells as tumor-resident Tregs and PD-1 as a suppressor.
ATLL clones were infected prior to T-cell receptor gene rearrangement revealing a previously unappreciated origin from T-cell precursors.

Abstract
Immune checkpoint inhibitors are a powerful new tool in the treatment of cancer, with prolonged responses in multiple diseases, including hematologic malignancies, such as Hodgkin lymphoma. However, in a recent report, we demonstrated that the PD-1 inhibitor nivolumab led to rapid progression in patients with adult T-cell leukemia/lymphoma (ATLL) (NCT02631746). We obtained primary cells from these patients to determine the cause of this hyperprogression. Analyses of clonality, somatic mutations, and gene expression in the malignant cells confirmed the report of rapid clonal expansion after PD-1 blockade in these patients, revealed a previously unappreciated origin of these malignant cells, identified a novel connection between ATLL cells and tumor-resident regulatory T cells (Tregs), and exposed a tumor-suppressive role for PD-1 in ATLL. Identifying the mechanisms driving this alarming outcome in nivolumab-treated ATLL may be broadly informative for the growing problem of rapid progression with immune checkpoint therapies.
Introduction
Checkpoint inhibitors are rapidly changing the management of cancer, with high rates of clinical response in multiple diseases, including renal cell carcinoma, metastatic melanoma, and Hodgkin lymphoma.1-3 However, accelerated tumor progression after anti–PD-1 therapy has been reported in a subset of patients.4,5 This finding highlights the critical need to understand the mechanism of hyperprogression with the use of these novel agents in multiple disease settings.
Adult T-cell leukemia/lymphoma (ATLL) is an important model system to interrogate this problem. ATLL is a malignancy of mature CD4+ T cells that occurs in 2% to 5% of people infected with a retrovirus, human T-cell leukemia virus-1 (HTLV-1).6 ATLL presents as smoldering, chronic, acute, and lymphoma subtypes, which are generally resistant to therapy. Irrespective of the clinical subtypes, ATLL is characterized by a very poor prognosis.7 Because of the endemic pattern of HTLV-1, ATLL is most often diagnosed in Japan, the Caribbean region, and Latin America. Genomic analyses of Japanese ATLL have demonstrated a high frequency of mutations, including gain-of-function mutations in genes encoding components of the T-cell receptor (TCR) activation pathway and mutations in immune surveillance genes, as well as high levels of PD-L1 expression.8 Most ATLL patients diagnosed in North America are of Caribbean descent and appear to have a somewhat different mutational signature.9 Yet, the clinical significance of such differences is unknown. Based on the involvement of the PD-1/PD-L1 axis in ATLL pathogenesis, we initiated a multicenter single-arm phase 2 trial of the PD-1 inhibitor nivolumab for subjects with ATLL; however, this clinical trial was discontinued after the first 3 patients enrolled in the study unexpectedly developed rapid progression of disease after a single infusion.10
ATLL cells are typically CD4+ and CD25+ and have characteristics similar to regulatory T cells (Tregs).11,12 Tregs are a subset of suppressor T cells that are critically involved in peripheral tolerance, inhibition of effector T cells, and suppression of autoimmunity. PD-1 is expressed on Tregs and partially regulates Treg generation and function.13 Tissue-resident Tregs have a somewhat different gene expression pattern compared with Tregs in the peripheral blood. Tumor-associated Tregs are a unique subset of tissue-resident Tregs.14 They often express factors that regulate lymphocyte activation, such as CD27, CTLA4, ICOS, GITR, OX40, and TIGIT, as well as other genes like MAGEH1, CCR8, and CD177.15 The functional effects of Tregs on tumor progression are context dependent, promoting tumor progression in hepatocellular carcinoma by suppressing tumor immunity while inhibiting progression of colorectal carcinoma by suppressing inflammation.16 Here we present data that indicate a suppressive role for PD-1 in indolent ATLL, and we report the discovery of a similar gene-expression profile between tumor-associated Tregs and ATLL cells after PD-1 blockade. We also document a clonal composition change following PD-1 blockade, and explore mechanisms that may explain the rapid progression of disease in ATLL patients upon nivolumab treatment.
Methods
Clinical samples
Peripheral blood mononuclear cells (PBMCs) were isolated and viably frozen from whole blood collected during the course of treatment from 3 patients, as described.10 Patient 2 refused consent for additional blood samples to be obtained after nivolumab treatment. The clinical study sites’ institutional review boards or ethics committees approved this study. All patients provided written informed consent.
Clonality analysis and sequencing
Analysis of hybrid capture DNA sequence data, using probes for HTLV-1 sequences and recurrently mutated cancer genes, was performed as described in the supplemental Methods (available on the Blood Web site).
In vitro T-cell proliferation assay
All test samples were maintained in Iscove modified Dulbecco medium supplemented with 20% human serum and 100 U/mL Recombinant Human IL-2 (cat. no. 200-02; PeproTech). Ninety-six–well round-bottom plates were coated with anti-human CD3 (3 μg/mL; clone OKT3; cat. no. 317315; BioLegend) and control human immunoglobulin G or human PD-L1–Ig (10 μg/mL; cat. no. 110-HG-100 and 156-B7-100; R&D Systems) in 50 μL of phosphate-buffered saline per well overnight at 4°C. Plates were washed with phosphate-buffered saline, and ATLL cells were seeded in duplicates at 5 × 104 cells per 100 μL of medium. The total number of viable cells was measured 3 days later using a Cell Counting Kit - SK (cat. no. CK10; Dojindo), according to the manufacturer’s instructions.
Flow cytometry
Flow cytometry, immunohistochemical, and pathologic analyses of primary tissue specimens were performed in the flow cytometry and surgical pathology clinical laboratories of the National Institutes of Health, Montefiore Medical Center, Albert Einstein College of Medicine, and The Ohio State University. Antibodies used for additional flow cytometry analysis after short-term culture included FITC anti-human PD-1 (clone EH12.2H7; cat. no. 329904) and APC anti-human PD-L1 (clone 29E.2A3; cat. no. 329708; both from BioLegend).
Reverse phase protein array analysis
Cell lysates of PBMCs obtained from patient 1 were submitted to the Functional Proteomics Reverse Phase Protein Array Core facility at MD Anderson Cancer. Lysates were serially diluted and arrayed on nitrocellulose-coated slides, probed with 301 antibodies using a tyramide-based signal amplification approach, visualized by DAB colorimetric reaction, and scanned on a Huron TissueScope scanner; spots were identified and densities were quantified using an Array-Pro Analyzer. Relative protein levels for each sample were determined by interpolating each dilution curve produced from the densities of the serially diluted sample spots using a standard curve for each antibody. All relative protein level data points were normalized for protein loading and transformed to linear values.
RNA sequencing
Total RNA was obtained from PBMCs from patient 1 and patient 3 before and after nivolumab treatment using RNeasy (QIAGEN). RNA was submitted to the Genome Technology Access Center at Washington University for RNA sequencing (RNAseq) analysis processed with Ribo-Zero for removal of ribosomal RNA and sequenced on a HiSeq 2500 using 1 × 50 paired-end run. Reads were aligned to the reference genome using STAR, and gene counts were derived from uniquely aligned reads using Subread:featureCount.17,18 BAM files for RNAseq and probe capture DNA were submitted to the Sequence Read Archive Database under BioProject accession number PRJNA555621.
Analysis of GSE33615 microarray data
Data from an ATLL microarray19 were downloaded from the National Center for Biotechnology Information Gene Expression Omnibus (accession number GSE33615). In the original study, RNA was extracted from PBMCs isolated from patients with acute ATLL (n = 26), chronic ATLL (n = 20), lymphomatous ATLL (n = 1), and smoldering ATLL (n = 4) and compared with RNA obtained from CD4+ cells from 21 normal subjects. In this study, values were normalized to actin (ACTB) and then represented as foldincrease compared to the value for patient 10 (a smoldering ATLL sample with the lowest proviral load in the study).
Mouse models of ATLL
Results
Nivolumab resulted in rapid expansion of predominant ATLL clones
All 3 patients enrolled in clinical trial NCT02631746 experienced rapid expansion of disease after 1 infusion of nivolumab, as described in the supplemental Clinical information.10 Genomic DNA purified from PBMCs obtained from 3 ATLL patients before nivolumab therapy, as well as from 2 of the 3 patients after therapy, was sequenced using a probe-capture next-generation method for detection of proviral integration sites and somatic mutations commonly associated with ATLL. The same DNA samples were also analyzed for TCR gene rearrangements of the TRB and TRG loci (Invivoscribe). Prior to therapy, all 3 patients had highly clonal disease, with a single predominant proviral integration site representing 63%, 98% (data not shown), and 81% of reads obtained (Figure 1). In the 2 evaluable cases, both predominant clones expanded after a single dose of nivolumab.
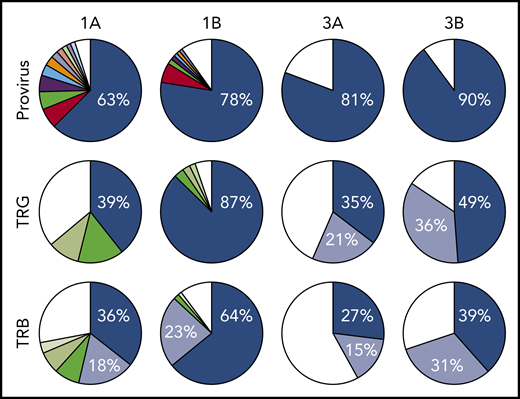
Nivolumab treatment resulted in rapid expansion of predominant ATLL clones. Clonal analysis of the peripheral blood of patients 1 and 3 before (A) and after (B) nivolumab therapy comparing probe-capture sequencing for HTLV-1 proviral integration sites with TCR gene rearrangement of β locus (TRB; supplemental Table 2) and γ locus (TRG; supplemental Table 1). Clones < 1% of total reads are shown in white.
Nivolumab treatment resulted in rapid expansion of predominant ATLL clones. Clonal analysis of the peripheral blood of patients 1 and 3 before (A) and after (B) nivolumab therapy comparing probe-capture sequencing for HTLV-1 proviral integration sites with TCR gene rearrangement of β locus (TRB; supplemental Table 2) and γ locus (TRG; supplemental Table 1). Clones < 1% of total reads are shown in white.
Clones that rapidly expanded after PD-1 blockade carry mutations commonly found in ATLL
Targeted exon sequencing was performed to identify alterations in the 39 genes most commonly mutated in ATLL,8 at a read depth for most genes between 5000× and 10 000× coverage, resulting in a sensitivity limit of detecting a variant allele frequency as low as 0.05% of total reads (Figure 2). A NOTCH1 mutation, found in patient 3 after nivolumab, was the only mutation not detected prior to treatment. DNA from patient 3 was also analyzed using the NexCourse Complete platform (Genoptix), which interrogates 236 genes recurrently mutated in hematologic and solid tumor neoplasms at a read depth of ∼500× coverage, with a limit of detection of 5% for single nucleotide variations. Sixteen genes were included in both sequencing methodologies, but 3 of the 4 mutations detected by the probe capture method were not evaluated in the NexCourse Complete pipeline. The NOTCH1 mutation was confirmed on the NexCourse Complete platform, and no additional mutations were identified. Patient 2 and patient 3 shared the D427N mutation in PRKCB, which is the most common recurrent amino acid substitution found in ATLL.9
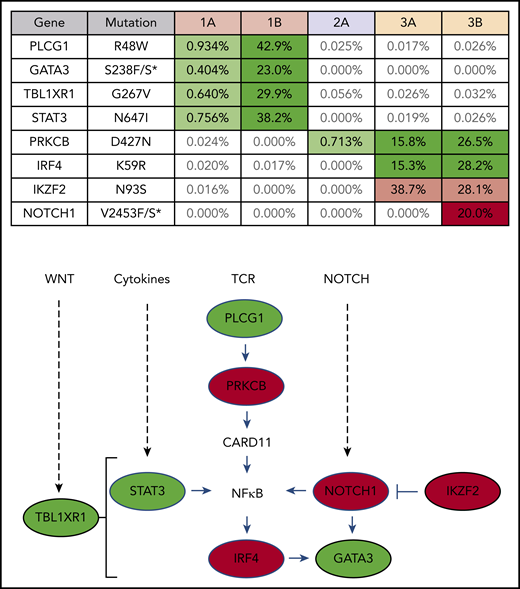
ATLL clones that rapidly expanded after PD-1 blockade carry mutations commonly found in ATLL. Mutations present in patient PBMCs before (A) and after (B) nivolumab therapy. Percentage of reads is for mutant over total number of reads for each gene segment. Mutations that increased in abundance after nivolumab are shaded green, the mutation that decreased after nivolumab is shaded pink, and the mutation present only after nivolumab is shaded red in the table. The schematic diagram below the table shows the role of each gene product in T-lymphocyte signaling pathways (green, mutations in patient 1; red, mutations in patient 3).
ATLL clones that rapidly expanded after PD-1 blockade carry mutations commonly found in ATLL. Mutations present in patient PBMCs before (A) and after (B) nivolumab therapy. Percentage of reads is for mutant over total number of reads for each gene segment. Mutations that increased in abundance after nivolumab are shaded green, the mutation that decreased after nivolumab is shaded pink, and the mutation present only after nivolumab is shaded red in the table. The schematic diagram below the table shows the role of each gene product in T-lymphocyte signaling pathways (green, mutations in patient 1; red, mutations in patient 3).
Predominant ATLL clones originated from infected immature T cells and rapidly expanded after nivolumab treatment
Analysis of 3 independent measures of clonality, proviral integration and TRG and TRB rearrangements, revealed an unexpected commonality among all 3 patients. In each patient, different TCR clones shared the same proviral integration site, indicating that HTLV-1 infection occurred in an immature T-cell precursor prior to completion of TCR gene rearrangements (Figure 1). TCR and sequence data, combined with flow cytometry data obtained from blood and tissue biopsies (supplemental Figure 1), suggested that, for patient 1, viral infection of an immature T cell resulted in expansion of a T-cell clone with a proviral integration site on chromosome 20, a Vg10-JgP1 TRG rearrangement (supplemental Table 1). This clone also carried an R48W mutation in PLCG1, which likely occurred early in the clonal evolution process (Figure 2). This clone subsequently split into 2 subclones, each with a different TRB rearrangement (supplemental Table 2). Both subclones were CD25+ CD45+ CD52+ CD5+ CD8−, but 1 subclone was CD4+ and the other was CD4− (supplemental Figure 1).
In patient 2, who presented with smoldering ATLL charpacterized by atypical CCR8+ CD4+ CD8− CD25+ PD-1− cells, 2 predominant TRG clones with identical proviral integration sites in chromosome 2 were detected in the peripheral blood. Similarly, infection of an immature T cell in patient 3 resulted in expansion of a malignant clone that produced 2 subclones with identical proviral integration sites but unique TCR gene rearrangements. The primary subclone in the peripheral blood (TRG Vg2; TRB Vb5-1, Jb2-3) expressed Tax and HBZ and carried an IKZF2 N93S mutation. The secondary clone (TRG Vg3 Jg1/2; TRB Vb19, Jb2-1) carried IRF4 K59R and PRKCB D427N mutations and expressed HBZ but not Tax. This is based on the finding that proviral load increased 2.4-fold in this patient, and HBZ RNA increased 2.1-fold, but Tax expression decreased by 20%, and IKZF2 copy number decreased by 23% (Figure 2; data not shown).10 The fact that the cell that expanded into a malignant clone in all 3 ATLL patients was initially infected as an immature T cell may be relevant in understanding the origin of ATLL cells and their rapid expansion after nivolumab therapy.
After PD-1 blockade, circulating ATLL cells expressed genes characteristic of tumor-infiltrating Tregs
Prior to therapy, 2 dominant ATLL subclones were present in patient 1; both were CD5+ CD8− CD25+ CD45+ CD52+, and both carried the same proviral integration site (Figure 1); however, 1 was CD4+, and the other was CD4− (supplemental Figure 1). Both clones could be detected in the peripheral blood, but the CD4− CD8− clone found primarily in the spleen and skin was also PD-1− and PD-L1−. Nivolumab treatment resulted in dramatic expansion of the double-negative clone in the peripheral blood, with concomitant expression of genes associated with tumor-associated Tregs, including MAGEH1, CCR8, CTLA4, CD27, CD70, TNFRSF4, and LAG-3 (Figure 3).15 Moreover, these abnormal cells in the peripheral blood and skin lesion expressed high levels of PD-1 after nivolumab therapy but remained PD-L1−.
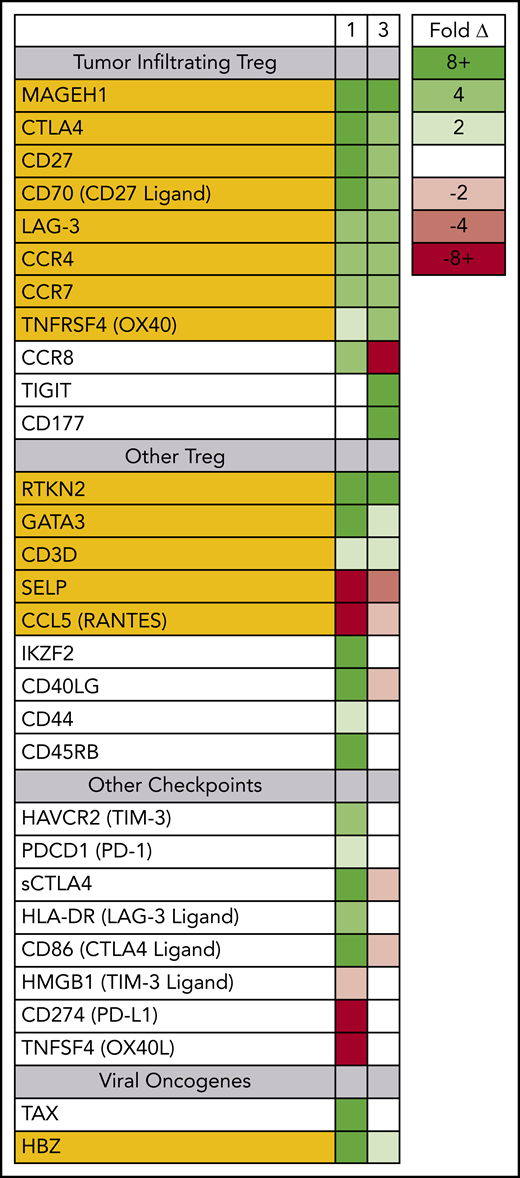
Circulating ATLL cells posttreatment resemble tumor-infiltrating Tregs. RNAseq was used to profile gene-expression changes caused by nivolumab treatment. The color bar indicates the fold change (after/before) for each gene. Selected genes represent genes associated with Tregs and lymphocyte checkpoint pathways that increase or decrease more than twofold after treatment with nivolumab.
Circulating ATLL cells posttreatment resemble tumor-infiltrating Tregs. RNAseq was used to profile gene-expression changes caused by nivolumab treatment. The color bar indicates the fold change (after/before) for each gene. Selected genes represent genes associated with Tregs and lymphocyte checkpoint pathways that increase or decrease more than twofold after treatment with nivolumab.
The pretreatment bone marrow from patient 2 showed PD-1 staining in 10% to 20% of lymphocytes, although an earlier skin biopsy from the same patient was negative for PD-1.
For patient 3, prior to nivolumab therapy, a bone marrow biopsy showed atypical T cells expressing CD2, CD3, CD4, CD5, and CD25, but not CD7 or CD8, and there were few atypical T cells detected in the peripheral blood. After nivolumab treatment, the patient experienced new splenomegaly and worsening lymphadenopathy, and 30% of nucleated cells in the peripheral blood were atypical “flower” lymphoid cells, with a similar phenotype as that noted in the bone marrow. Similar to patient 1, PD-1 blockade was associated with increased expression of genes associated with tumor-associated Tregs,15,23 including MAGEH1, CTLA4, CD27, CD70, TNFRSF4, and LAG-3. However, unlike patient 1, the expanding ATLL cells expressed PD-L1 and had limited expression of CCR8 and PD-1 (supplemental Figure 2). Both evaluable patients, each carrying clones with different driver mutations, experienced rapid progression after nivolumab, and the rapidly expanding circulating ATLL cells had increased expression of genes reported to be characteristic of tumor-associated Tregs.
ATLL clones that expanded in 2 patients after nivolumab represent distinct Treg subtypes
Although the rapidly expanding ATLL clones in both evaluable patients expressed genes seen in tumor-associated Tregs, the 2 patients also had signatures of very different Treg subtypes (Figure 4). In patient 1, the rapidly expanding CCR8+ CD4− CD25+ PD-1+ PD-L1− ATLL cells in the peripheral blood were associated with increased expression of the soluble isoform of CTLA4, CD86, CD40LG, CD45RB, granzymes A, B, and H, both viral oncogenes (Tax and HBZ), and genes associated with T-cell activation, including CD3E, IL2, LFA-1 (both ITGB2 and ITGAL), ICAM3, and IL10. The cells in the peripheral blood after nivolumab treatment also exhibited reduced expression of genes associated with innate immunity (LTF, DEFA4, DEFA3, LCN2, BPI), platelet granules (PF4, PPBP, CLU, TLN1), and interleukin-8 (IL-8) signaling (CXCL8, CXCR1, CXCR2, IL1B), as well as a dramatic reduction in FAK signaling (PTK2, CCL5, TNFSF10, IL1RN, CCL7, CCL2, CCL4) (supplemental Figure 3).24 This gene-expression pattern is indicative of the characteristics and activity of CCR8+ PD-1+ Tregs, which have been described as master drivers of immunosuppression.25
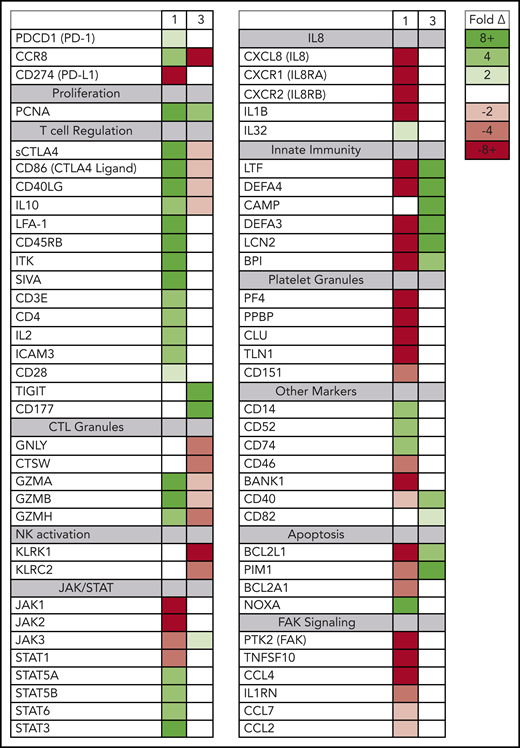
Posttreatment ATLL cells in patients 1 and 3 represent distinct Treg subtypes. RNAseq was used to profile gene-expression changes caused by nivolumab treatment. The color bar indicates the fold change (after/before) for each gene. Selected genes represent genes associated with peripheral blood cell signaling pathways that increase or decrease more than twofold after treatment with nivolumab and differ between patient 1 and patient 3.
Posttreatment ATLL cells in patients 1 and 3 represent distinct Treg subtypes. RNAseq was used to profile gene-expression changes caused by nivolumab treatment. The color bar indicates the fold change (after/before) for each gene. Selected genes represent genes associated with peripheral blood cell signaling pathways that increase or decrease more than twofold after treatment with nivolumab and differ between patient 1 and patient 3.
In contrast, in patient 3 the rapidly expanding CCR8− CD4+ CD25+ PD-1− PD-L1+ cells correlated with decreasing levels of CCR8, soluble CTLA4, and CD86; no or very low expression of PD-1; and increased expression of CD177, TIGIT, CD40, and BCL2L1. Also unlike patient 1, nivolumab treatment resulted in activation of defensins and innate immunity (LTF, DEFA4, CAMP, DEFA3, LCN2, BPI), decreased expression of genes related to cytotoxic T lymphocyte granules (GNLY, CTSW, GZMA, GZMB, GZMH), and decreased expression of natural killer cell markers (KLRK1, KLRC2). This gene-expression pattern is consistent with CCR8− PD-1− Tregs, which have previously been reported to uniquely activate defensins.26 Taken together, these data suggest that rapid ATLL expansion after PD-1 blockade is associated with genes that are usually activated in tumors and independent of the unique subtype of the atypical cell present in each patient.
Discussion
Recently, there have been reports of rapid progression occurring in a small minority of cancer patients after PD-1 blockade that must be recognized as part of the landscape for this highly promising therapeutic option. Although the sample size in our study is small, all of the ATLL patients treated with nivolumab had rapid and dramatic progression of disease. Our analyses have revealed acute dysregulation of gene function in these patients, including the appearance of unique molecular signals, but no unifying pathophysiology that could explain these events. Nevertheless, it is clear from these patients’ experiences that a better understanding of the factors and mechanisms that govern this harmful outcome is urgently needed. In our ATLL patients who experienced such a response to nivolumab, the expansion of all predominant clones carrying a variety of mutations suggested that rapid expansion of ATLL after PD-1 blockade reflected an unanticipated loss of ATLL suppression rather than a selective advantage for a specific clone. Unexpectedly, the clonal analysis also exposed the fact that the ATLL clones were infected by HTLV-1 prior to completion of TCR gene rearrangements.27 However, recent work has shown that HTLV-1 can infect hematopoietic precursors, in addition to mature lymphocytes.28 Only a small percentage of people infected with HTLV-1 develop ATLL, whereas the vast majority remain asymptomatic for life. It remains unclear whether infection and clonal expansion of T-cell precursors are related to viral latency or pathogenesis or are a prerequisite for ATLL.
In these cases of ATLL, the atypical cells that expanded in the peripheral blood after nivolumab treatment expressed genes characteristic of tumor-associated Tregs. Nivolumab is known to shift the profile of circulating Tregs toward an exhausted phenotype.23 However, in ATLL, nivolumab seemed to remove suppression of tissue-resident ATLL cells, causing expansion in the involved tissue and into the peripheral blood. Several mechanisms may be involved in this phenotype, including compensatory upregulation of additional checkpoints, loss of PD-1–mediated tumor suppression in the malignant T cells, and/or loss of a PD-1–mediated immune response common to indolent ATLL.
The similarities in gene expression among the ATLL cells in these patients and different Treg-like subtypes provide more support for the hypothesis that ATLL is a Treg malignancy and suggest that the rapidly developing field of Treg biology could help to clarify disparate findings that persist in HTLV research. For example, understanding the difference between CCR8− PD-1− Tregs that activate defensins vs CCR8+ PD-1+ Tregs that drive immunosuppression may reveal a corollary among subtypes of ATLL. Moreover, the expression of multiple checkpoints on tumor-associated Tregs also seems to be applicable to aggressive forms of acute and lymphomatous ATLL. Finally, CD4− CD8− ATLL clones, like the one that expanded in patient 1, may have potent suppressive characteristics similar to double-negative Tregs.29,30
Mouse models of aggressive ATLL do not recapitulate rapid progression seen in these patients
NSG-Kit mice, humanized by intrahepatic injection of CD34+ cells freshly isolated from cord blood and infected with HTLV-1 after 6 to 12 weeks, typically develop an aggressive oligoclonal lymphoproliferative disease 4 to 12 weeks postinfection.20 However, nivolumab treatment in this model resulted in decreased numbers of human T cells in the blood and spleen after 5 weeks (supplemental Figure 4). Analogous results were recently reported in a model of infected NSG mice, humanized with CD133+ hematopoietic cells.31 Similarly, a PDX model in which PBMCs, freshly obtained from a patient with acute ATLL, were injected intraperitoneally into NSG-Kit mice typically results in expansion of the ATLL graft and death within 8 to 12 weeks.22 Nivolumab treatment in the PDX model blocked ATLL proliferation and decreased organ and blood involvement (supplemental Figure 4). The transgenic model in which Tax is driven by the granzyme B promoter produces spontaneous tumors between 6 and 9 months of age.21 PD-1 blockade in tumor-bearing Tax mice resulted in decreased tumor growth (supplemental Figure 4). In each of these mouse models of ATLL, PD-1 blockade resulted in decreased disease rather than rapid progression. In response to our original study, investigators from Japan shared their nivolumab trial experience; their ATLL patients generally do not experience rapid progression after nivolumab treatment.32 Taken together, this suggests that there exists a subset of ATLL patients or a stage or subtype of ATLL that can be exacerbated by nivolumab that is not represented in mouse models of acute disease.
Gene-expression changes after nivolumab support several potential mechanisms
PD-1 plays an important role in holding tumor-resident ATLL cells in check, independently of whether the cells express the ligand or the receptor. Several mechanisms, which are not mutually exclusive, could explain the unexpected outcome that nivolumab accelerated disease in all 3 ATLL patients (Figure 5).
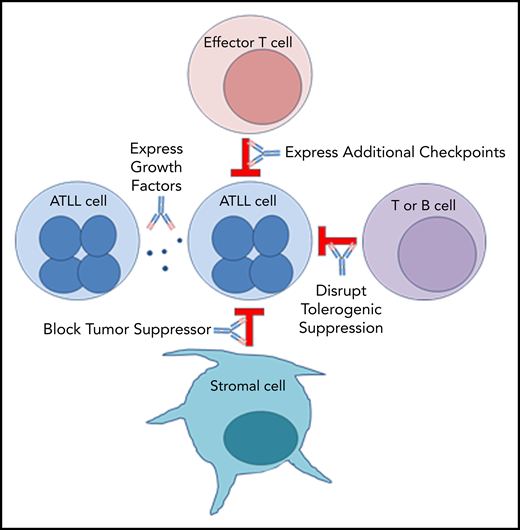
Mechanism 1: PD-1 blockade drives expression of additional checkpoints that promote the suppressive activity of ATLL cells
In both cases, expanding ATLL clones expressed increasing levels of CTLA4, LAG-3, and OX40, as well as TIM-3, ICOS, and PD-1 in patient 1 and TIGIT and CD40 in patient 3. Compensatory upregulation of additional checkpoints has been described as an escape mechanism of PD-1 blockade escape. CTLA4, LAG-3, and TIGIT are frequently overexpressed in ATLL cells along with CD27 and CD70 (supplemental Figure 5). CD27 increases Treg proliferation, protects Tregs from apoptosis, and is associated with suppressive Treg activity. In the 6 months prior to the start of treatment, patient 1 began to shift from classic CD3low CD4+ CD25+ ATLL cells with the emergence of a new CD3low CD4−CD8− population. This was the dominant ATLL clone after treatment, and posttreatment analysis of a skin lesion showed that 68% of T cells were CD3low CD4− CD8− CD25+ PD-1+ PD-L1− ATL cells, whereas stroma and macrophages were PD-L1+. Kamada et al recently described hyperprogression in advanced gastric cancer in which nivolumab augmented the proliferation and suppressive activity of Tregs, which then overwhelmed the tumor-reactive cytotoxic T-lymphocyte response.33 The expression of these additional checkpoints following PD-1 blockade may facilitate the escape of the rapidly expanding clones.
Mechanism 2: PD-1 blockade alters expression of ATLL-promoting growth factors
In patient 1, PD-1 blockade was associated with an increase in IL-2 and IL-10, both of which promote proliferation.34,35 Moreover, reduced expression of IL-8, a T-cell chemoattractant that is often expressed in chronic viral infections, could block recruitment of potent CXCR1+ CD8 T cells.36 Similarly, reduced expression of FAK, an inhibitor of TCR signaling, could promote ATLL expansion.37
Similar expression patterns were not detected in patient 3; however, after nivolumab treatment, a population of ATLL cells with a mutation in the PEST domain of Notch1 was identified in patient 3 (Figure 1). Similar mutations were described as activating mutations in 15% to 30% of ATLL cases, resulting in increased stability of the Notch1 protein and enhanced proliferation of ATLL cells.8,38-40
Mechanism 3: Stromal PD-L1 acts as a tumor suppressor by binding to PD-1 expressed on leukemic cells in the ATLL microenvironment
PD-1 was identified as a potent tumor suppressor in the context of a T-cell malignancy driven by constitutive TCR signaling, and its inhibition is hypothesized to increase costimulatory CD28 signaling and the downstream phosphatidylinositol 3-kinase/AKT pathway.41,42 However, expression of PTEN, PRKCQ, and AKT1 remained unaffected by nivolumab treatment in patients 1 and 3. On the other hand, CD28, STAT3, STAT5A/B, and STAT6 were elevated after nivolumab in patient 1. In patient 3, the postrelapse cells, but not those prior to nivolumab treatment, were responsive to growth inhibitory control by PD-L1 in an in vitro setting (supplemental Figure 2). These observations are consistent with the notion that PD-L1–expressing cells in the microenvironment suppress PD-1+ ATLL cells and that nivolumab neutralized this suppressive function, leading to clonal expansion of PD-1+ ATL cells.43
Mechanism 4: PD-1 uniquely governs immunity in patients with chronic or smoldering ATLL
ATLL patients with smoldering or chronic disease often have increased expression of TCL1A, MS4A1, EBF1, IGHM, CD22, CD19, CD40, and BANK1 in their PBMCs compared with normal patients and patients with acute ATLL (supplemental Figure 6). BANK1 was also highly expressed in patient 1 before PD-1 blockade. Interestingly, these markers are also associated with operational tolerance as opposed to acute rejection in human allogeneic transplantation. Patients who have higher levels of these genes achieve operational tolerance and no longer require antirejection immunosuppressive medication to prevent graft rejection.44 The fact that PD-1 blockade affected the expression of a gene associated with operational tolerance suggests that PD-1 may be mediating an immune condition in which T-cell proliferation is restricted in stable forms of ATLL and in operational tolerance after transplantation. The rapid proliferation of ATLL after PD-1 blockade may suggest that aggressive vs indolent forms of ATLL are not intrinsic to the ATLL cell but are a characteristic of the immune response in that subset of patients. This mechanism may also explain the rapid expansion of infected clones in immune-suppressed organ transplant recipients who received organs from HTLV-1–infected donors.45
In conclusion, taken together, these data reveal a previously unappreciated origin of ATLL cells, confirm the report of rapid expansion of ATLL after PD-1 blockade in these patients, identify a novel connection between ATLL cells and tumor-resident Tregs, and expose an unresolved suppressive role for PD-1 in ATLL. Development of a model system that faithfully recapitulates these aspects of ATLL will be essential for establishing the mechanism underlying these findings and whether this is more broadly informative to the alarming problem of rapid progression of malignancy after immune checkpoint therapy. Furthermore, although primarily hypothesis generating, these results demonstrate the critical need to understand the underlying mechanism of immune therapies as we apply these promising treatments to an ever-increasing scope of diseases.
BAM files for RNAseq and probe capture DNA were submitted to the Sequence Read Archive Database under BioProject accession number PRJNA555621.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Matthew Cherian, Zhangfang Guo, and Cynthia Ma for assistance with processing samples, Sarah Larson and Christy Arrowood for coordinating the study, and Howard Streicher for helpful suggestions. The authors also thank Invivoscribe, McDonnell Genome Center, Siteman Cancer Center Genomic Technology Access Core, Genoptix, and the Functional Proteomics Reverse Phase Protein Array Core Facility at MD Anderson Cancer Center for clinical data. The authors especially thank the patients and support staff who participated in clinical trial NCT02631746.
This work was supported by Public Health Service grants UM1 186707, CA23460, CA063417, CA100730, CA016672, and HG007940; funds from the Siteman Cancer Center and intramural National Cancer Institute Program; and a grant from the Dr. Louis Skarlow Memorial Trust (B.H.Y.).
Authorship
Contribution: D.A.R., J.C.H., B.H.Y., X.Z., X.R., S.O., X.C., H.S., and A.J. performed laboratory experiments; K.C.C., M.J., J.E.B., M.D.M., and T.A.W. provided clinical samples and data; D.A.R., B.H.Y., Z.L.S., O.G, and M.G. performed informatics studies; L.R. conceived the clinical and experimental studies; and all authors reviewed and approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Lee Ratner, Washington University, 660 S Euclid Ave, Box 8069, St. Louis, MO 63110; e-mail: lratner@wustl.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal