

Key Points
Fibronectin disrupts PF4/heparin complexes and inhibits anti-PF4/heparin antibody–mediated platelet activation in vitro.
Low plasma fibronectin is associated with increased anti-PF4/heparin antibody formation and increased breakthrough of HIT.

Abstract
Heparin-induced thrombocytopenia (HIT) is caused by platelet-activating anti–platelet factor 4 (PF4)/heparin antibodies. Platelet activation assays that use “washed” platelets are more sensitive for detecting HIT antibodies than platelet-rich plasma (PRP)–based assays. Moreover, heparin-exposed patients vary considerably with respect to the risk of PF4/heparin immunization and, among antibody-positive patients, the risk of subsequent “breakthrough” of clinical HIT with manifestation of thrombocytopenia. We used washed platelets and PRP, standard laboratory HIT tests, and physicochemical methods to identify a plasma factor interfering with PF4/heparin complexes and anti-PF4/heparin antibody–platelet interaction, thus explaining differences in functional assays. To investigate a modulating risk for PF4/heparin immunization and breakthrough of HIT, we also tested 89 plasmas from 2 serosurveillance trials. Fibronectin levels were measured in 4 patient groups exhibiting different degrees of heparin-dependent immunization and expression of HIT. The heat-labile plasma protein, fibronectin, inhibited PF4 binding to platelets in a dose-dependent fashion, particularly in washed (vs PRP) systems. Fibronectin also inhibited PF4/heparin binding to platelets, anti-PF4/heparin antibody binding to PF4/heparin complexes, and anti-PF4/heparin antibody–induced platelet activation as a result of PF4/heparin complex disruption. In addition, plasma fibronectin levels increased progressively among the following 4 patient groups: enzyme-linked immunosorbent assay (ELISA)+/serotonin-release assay (SRA)+/HIT+ < ELISA+/SRA+/HIT− ∼ ELISA+/SRA−/HIT− < ELISA−/SRA−/HIT−. Altogether, these findings suggest that fibronectin interferes with PF4/heparin complex formation and anti-PF4/heparin antibody–induced platelet activation. Reduced fibronectin levels in washed platelet assays help to explain the greater sensitivity of washed platelet (vs PRP) assays for HIT. More importantly, lower plasma fibronectin levels could represent a risk factor for PF4/heparin immunization and clinical breakthrough of HIT.
Introduction
Heparin-induced thrombocytopenia (HIT) is a prothrombotic adverse drug effect1 caused by immunoglobulin G (IgG) antibodies directed against platelet factor 4 (PF4)/heparin complexes.2,3 Anti-PF4/heparin IgG trigger platelet activation via cross-linking of platelet FcγRIIA,4-6 promoting strong platelet activation, thrombocytopenia, hypercoagulability, and high thrombotic risk.7,8
Only anti-PF4/heparin antibodies of IgG class are clinically relevant. High plasma levels of anti-PF4/heparin IgG are associated with a greater likelihood for the presence of platelet-activating antibodies,9-11 whereas high levels of total IgG appear to reduce the ability of anti-PF4/heparin IgG to induce platelet activation.4,12,13
Systematic serosurveillance studies of heparin-treated patients show that only a subset of patients who form platelet-activating anti-PF4/heparin antibodies develop clinically overt HIT.14-16 These observations raise the question of whether plasma factors influence clinical “breakthrough” of HIT (“breakthrough” refers here to the subset of HIT antibody-positive patients who manifest thrombocytopenia compared with those who are antibody positive but do not develop thrombocytopenia). We based our experimental approach on the well-established empirical observation that “washed platelet” assays for HIT are more sensitive than assays using platelet-rich plasma (PRP).17,18 The heparin-induced platelet activation (HIPA) test19 and the serotonin-release assay (SRA)20,21 are washed platelet assays used for detecting heparin-dependent platelet-activating antibodies. Notably, plasma factor levels are greatly reduced by washing of platelets vs PRP-based assays.22
Fibronectin is a large glycoprotein dimer occurring in 2 forms: insoluble cellular fibronectin as a component of the extracellular matrix and soluble plasma fibronectin as an abundant plasma constituent. Major functions of fibronectin include cell adhesion, bacteria–host interaction, and wound healing. Here, we identify low plasma fibronectin levels as a potential risk factor for anti-PF4/heparin antibody formation and developing clinical HIT. First, fibronectin inhibited the ability of HIT antibodies to activate platelets, via several mechanisms, including inhibition of PF4 and PF4/heparin binding to purified platelets, inhibition of antibody binding to PF4/heparin complexes by complex disruption, and inhibition of anti-PF4/heparin antibody-induced platelet activation. Second, we found a clear trend in fibronectin levels: they were lowest in patients with serologically confirmed and clinically confirmed HIT, higher in antibody-positive patients who did not develop clinical HIT, and highest in heparin-exposed, but nonimmunized, controls.
Methods
Preparation of plasma, plasma fractions, and platelets
Plasma was obtained by centrifugation of hirudinized (10 µg/mL, lepirudin; Pharmion, Hamburg, Germany) whole blood. Serum was prepared from clotted whole blood (Vacutainer SST II Advance; BD, Plymouth, United Kingdom) of healthy volunteers by centrifugation (1680g, 10 minutes, room temperature [RT]) and additional centrifugation of the supernatant (7000g, 5 minutes, RT). In several experiments, serum was also heat treated (56°C or 100°C, 30 minutes) and centrifuged (7000g, 5 minutes, RT) or depleted from IgG by incubating with protein G Sepharose 4 fast flow or depleted from fibronectin/vitronectin by incubating with heparin Sepharose (60 minutes, RT; GE Healthcare, Uppsala, Sweden) and centrifugation (twice; 120g, 30 seconds, RT). IgG was purified by protein G using standard methods (supplemental Methods, available on the Blood Web site).23
PRP, gel-filtered platelets (GFPs), and washed platelets were prepared as described (supplemental Methods).
The ethical boards of McMaster University and Greifswald University approved the use of patient serum/plasma and the use of platelets from volunteers for in vitro experiments.
Human anti-PF4/heparin antibodies
For in vitro studies, sera used for assessing the effects of fibronectin or vitronectin on anti-PF4/heparin antibody binding to PF4/heparin complexes and the effects of fibronectin on anti-PF4/heparin antibody-induced platelet activation were obtained from patients testing positive by PF4/heparin IgG enzyme–linked immunosorbent assay (ELISA) and by the HIPA test.
For the clinical association studies, we took advantage of 2 prospective studies performed at McMaster University consisting of postoperative orthopedic surgery patients.24,25 Blood samples were collected systematically (irrespective of thrombocytopenia occurrence), and platelet counts were conducted daily, thus allowing for the study of anti-PF4/heparin antibody formation (with or without platelet-activating properties), in association with the development of thrombocytopenia (serosurveillance studies). From these studies, we tested plasma samples from patients with serologically and clinically confirmed HIT (anti-PF4/heparin IgG ELISA+, SRA+, all with a >50% platelet count decrease bearing a temporal relationship with HIT antibody formation and 10/17 with thrombotic complications), as well as from 42 anti-PF4/heparin IgG ELISA+ controls without HIT (but in whom the SRA was positive in 18 patients and negative in 24 patients). In addition, 30 plasma samples were studied, with no anti-PF4/heparin antibodies detected by ELISA.26
Mass spectrometry
Mass spectrometry to detect proteins attached to serum-treated protein G Sepharose was performed as described (supplemental Methods).
Recombinant protein purification of fibronectin fragments
His6-tagged fibronectin fragments FNIII7-10 and FNIII12-14, consisting of the type III repeats 7, 8, 9, and 10 of human fibronectin (including the RGD cell binding domain) or type III repeats 12, 13, and 14 (including a heparin-binding domain) were recombinantly produced as described (supplemental Methods).
PF4 binding to platelets and plasma factors
PRP, GFPs, or washed platelets (40 000 per microliter) were incubated (30 minutes, 37°C) with increasing concentrations of human platelet–derived PF4 (ChromaTec, Greifswald, Germany) or suspension buffer, fixed (1% paraformaldehyde, 20 minutes, 4°C; Merck, Darmstadt, Germany), washed twice with suspension buffer (600g, 7 minutes, 4°C), incubated with a rabbit anti-human PF4 (Dianova) FITC labeled with a FluoReporter FITC Protein Labeling Kit (Molecular Probes, Eugene, OR), and mouse anti-human CD42a-PE (BD Biosciences), or isotype controls (30 minutes, 4°C), washed, and analyzed by flow cytometry (Cytomics FC 500; Beckman Coulter, Krefeld, Germany). Antibody binding was quantified by geometric mean fluorescent intensity (GMFI).
GFPs (40 000 per microliter) were also incubated (30 minutes, 37°C) with PF4 (25 µg/mL) in the presence of increasing concentrations of homologous plasma, serum (8.5%, 17%, 34%, 68%), and, for subsequent experiments, with a fixed concentration of heat-treated serum (20%), IgG-depleted serum (20%), IgG-depleted serum (20%) substituted with purified IgG according to the original IgG serum concentration, fibronectin/vitronectin-depleted serum (20%), fibronectin/vitronectin-depleted serum (20%) substituted with fibronectin (100 µg/mL) or vitronectin (50 µg/mL), or suspension buffer and then processed as described above, before cytometric analysis.
In another series of experiments, GFPs (40 000 per microliter) were incubated (30 minutes, 37°C) with PF4 (10 µg/mL) in the presence of increasing concentrations of α-2-macroglobulin (4-1000 µg/mL; Sigma-Aldrich), vitronectin (0.4-100 µg/mL; human plasma-derived; Merck Millipore, Darmstadt, Germany), or fibronectin (1.6-200 µg/mL; human plasma–derived; Invitrogen, Carlsbad, CA) or with 50 µg/mL fibronectin gelatin-binding fragment and fibronectin heparin-binding fragment (45 kDa and 30 kDa, respectively, both obtained by tryptic digestion of the N-terminal 70-kDa fragment; Sigma-Aldrich), fibronectin fragments 7 to 10 and fragments 12 to 14 of the type III domain, or suspension buffer and processed as described above, before cytometric analysis.
Influence of fibronectin and vitronectin on PF4/heparin complex binding to platelets
GFPs (40 000 per microliter) were incubated (30 minutes, 37°C) with PF4 (25 µg/mL) in the presence of increasing concentrations of unfractionated heparin (UFH; 150 IU/mg; Braun, Melsungen, Germany) and fibronectin (50 µg/mL), vitronectin (10 µg/mL), or suspension buffer concurrently, fixed, and processed as described above, before flow cytometric analysis.
Anti-PF4/heparin antibody assays
An in-house ELISA that detects anti-PF4/heparin IgG was used as described.27 In addition, increasing concentrations of UFH, fibronectin (100, 200, 400 µg/mL), or vitronectin (100, 200 µg/mL) were added to patient sera containing anti-PF4/heparin IgG prior to incubation with PF4/heparin coated onto a microtiter plate.
In addition, the amount of PF4 in the supernatant after incubating the PF4/heparin-coated plate with fibronectin was determined using a commercially available ELISA kit (Asserachrom; Diagnostica Stago, Asnières, France), according to the manufacturer’s instructions.
The HIPA test was performed, as described,18 using the low-molecular-weight heparin (LMWH) reviparin at 0.2 aFXaU/mL (reviparin induces greater maximal platelet activation in the presence of anti-PF4/heparin antibodies vs UFH).28 In addition to the optimal concentration of LMWH required for platelet activation, fibronectin (12.5, 25, 50, 100, 200 µg/mL) was added. Lag-time was monitored for 45 minutes (no activation = 45 minutes). In all functional experiments, high UFH concentrations (100 IU/mL) were used to show inhibition.
Additionally, washed platelets were incubated with fibronectin (200 µg/mL, 30 minutes, 37°C) or buffer and then washed (137 mM NaCl, 2.7 mM KCl, 12 mM NaHCO3, 0.4 mM NaH2PO4, 0.4% BSA, 0.1% glucose, 2.5 U/mL apyrase, 0.1 μg/mL hirudin [pH 6.3]; 600g, 7 minutes, 37°C) before use in the HIPA test, as described above.
Fibronectin interaction with PF4/heparin complexes using SPR and photon correlation spectroscopy
The direct protein–protein interactions between PF4/heparin complexes and fibronectin were assessed by surface plasmon resonance (SPR) using a Biacore T100 optical biosensor (GE Healthcare). Consumables, phosphate-buffered saline (PBS), and solutions for coupling and regeneration of the chip surface were purchased from GE Healthcare. Fibronectin was immobilized by a standard amine-coupling procedure on a carboxymethyl dextran (CM5) sensor chip. Briefly, 30 µg/mL fibronectin in 10 mM sodium acetate, pH 4.5, was coupled onto an N-hydroxysuccinimide (0.05 M)/N-ethyl-N′-(dimethylaminopropyl)carbodiimide (0.2 M)–activated sensor chip (70 µL, 10 µL/min), and the remaining reactive groups were deactivated by washing with 1 M ethanolamine, pH 8.5, for 7 minutes. Control flow cells were treated in the same way without the addition of protein. Binding of analytes was analyzed with serial dilutions of PF4/heparin complexes (40 µg/mL:1 U/mL) in PBS + 0.05% Tween, pH 7.4, at 25°C using a flow rate of 10 µL/min with an association time of 180 seconds and a dissociation time of 600 seconds in all experiments. Between subsequent sample injections, the surface was regenerated with 20 mM NaOH (20 seconds, 20 µL/min). Each protein interaction was carried out at least in triplicate and corrected for a blank injection without protein and reference channel interaction. Data analysis was carried out using BIAevaluation software (version 2.0.1).
Size measurements were carried out by photon correlation spectroscopy (PCS) using Zetasizer Nano ZS (green badge). The temperature was set to 25°C, and the measurement angle was 173°. The general purpose model was used for data analysis with Zetasizer software 6.12. The PF4/heparin complexes were formed by adding 0.5 U heparin to 20 µg of PF4 in 1 mL of PBS. Fibronectin was added to the solution to attain 3.1, 6.3, 12.5, 25, 50, 100, 200, and 400 µg/mL. Every concentration was observed for ≥30 minutes.
Determining fibronectin levels in patient plasma samples
Patient plasma obtained from the prospective studies in Hamilton, ON, Canada were analyzed for intact fibronectin levels by ELISA (American Diagnostica, Stamford, CT). Laboratory personnel were blinded to patient HIT status.
Statistical analysis
In vitro studies
Differences in PF4 binding of washed and GFPs compared with PRP and PF4 binding in the presence of heparin compared with heparin plus fibronectin or heparin plus vitronectin were calculated using analysis of variance. All other comparisons between groups were performed using the paired Student t test. P < .05 was considered statistically significant.
Clinical association studies
Fibronectin and anti-PF4/heparin IgG levels of anti-PF4/heparin ELISA− patients vs patients who formed anti-PF4/heparin antibodies and ELISA+ and SRA+ patients with clinical HIT vs ELISA+ and SRA+ but no clinical HIT patients were compared using the Wilcoxon rank-sum test. The fibronectin and anti-PF4/heparin IgG levels for all 4 groups were compared by trend (progressive expression of HIT), as follows: ELISA−/SRA−, ELISA+ but SRA−, ELISA+ and SRA+ but no clinical HIT, and ELISA+ and SRA+ with clinical HIT. The association between fibronectin and anti-PF4/heparin IgG levels was determined by median regression.
Results
Influence of plasma factors on PF4 binding to platelets
Platelets bound PF4 in a concentration-dependent manner. Binding of PF4 to washed platelets and GFPs saturated at ∼25 µg/mL PF4. GFPs (P < .0001 vs PRP) and washed platelets (P < .0001 vs PRP) showed a much higher binding capacity for PF4 than PRP (Figure 1A). The washing procedure is most likely more effective in removing plasma components than the gel-filtration method. Nevertheless, we used GFPs for the following experiments to avoid activation-related artifacts, because it is a more gentle method to separate platelets from plasma. Addition of plasma and serum to GFPs inhibited PF4 binding (25 µg/mL) in a dose-dependent manner starting at a concentration of 8.5% plasma (or serum) or higher (Figure 1B).
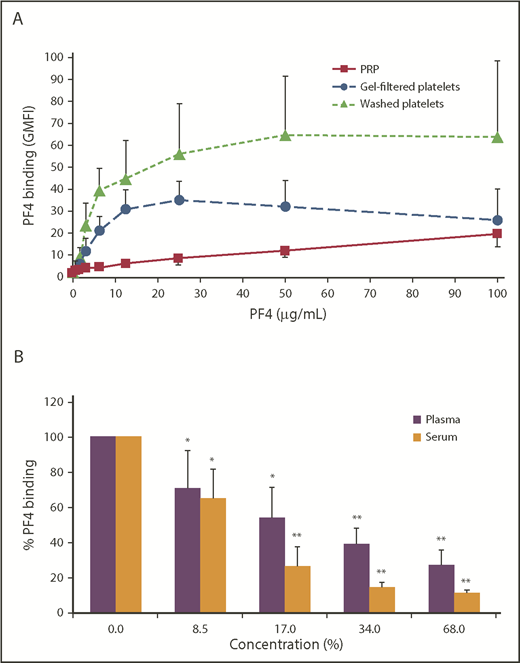
Plasma and serum inhibit PF4 binding to platelets. (A) PRP, GFPs, or washed platelets were incubated with increasing concentrations of PF4. PF4 binding was detected with a FITC-labeled anti-human PF4 antibody using flow cytometry, and antibody binding was quantified by GMFI. Data are mean ± standard deviation (SD) of 6 independent experiments. (B) GFPs were incubated with 25 µg/mL PF4 in the presence of increasing concentrations of plasma or serum. PF4 binding was detected with an FITC-labeled anti-human PF4 antibody using flow cytometry, and antibody binding was quantified by GMFI. Surface-bound PF4 without addition of plasma or serum was defined as 100%. Data are mean ± SD of 4 independent experiments. *P < .05, **P < .01 vs PF4 without plasma or serum.
Plasma and serum inhibit PF4 binding to platelets. (A) PRP, GFPs, or washed platelets were incubated with increasing concentrations of PF4. PF4 binding was detected with a FITC-labeled anti-human PF4 antibody using flow cytometry, and antibody binding was quantified by GMFI. Data are mean ± standard deviation (SD) of 6 independent experiments. (B) GFPs were incubated with 25 µg/mL PF4 in the presence of increasing concentrations of plasma or serum. PF4 binding was detected with an FITC-labeled anti-human PF4 antibody using flow cytometry, and antibody binding was quantified by GMFI. Surface-bound PF4 without addition of plasma or serum was defined as 100%. Data are mean ± SD of 4 independent experiments. *P < .05, **P < .01 vs PF4 without plasma or serum.
In the following experiments, we used a fixed serum concentration of 20%. Pretreatment at 100°C, but not at 56°C, resulted in loss of the inhibitory effect (Figure 2A). This excluded complement factors, which are inactivated at 56°C, and favored a proteinaceous nature of the inhibiting factor, because proteins are known to denature at 100°C, whereas polysaccharides are typically stable at that temperature. IgG-depleted serum obtained by affinity chromatography using protein G Sepharose no longer inhibited PF4 binding to GFPs; however, resubstitution of depleted serum with IgG did not restore inhibitory activity (Figure 2B). To assess whether the inhibitory factor binds to the Sepharose, we identified proteins (other than IgG) bound to the protein G Sepharose by mass spectrometry. The strongest signals were obtained for complement factors, α-2-macroglobulin, fibronectin, and vitronectin. Interestingly, pretreatment of serum with heparin Sepharose, which is used for purification of fibronectin and vitronectin, resulted in loss of inhibitory activity, and substitution with fibronectin or vitronectin restored inhibitory activity (Figure 2C).
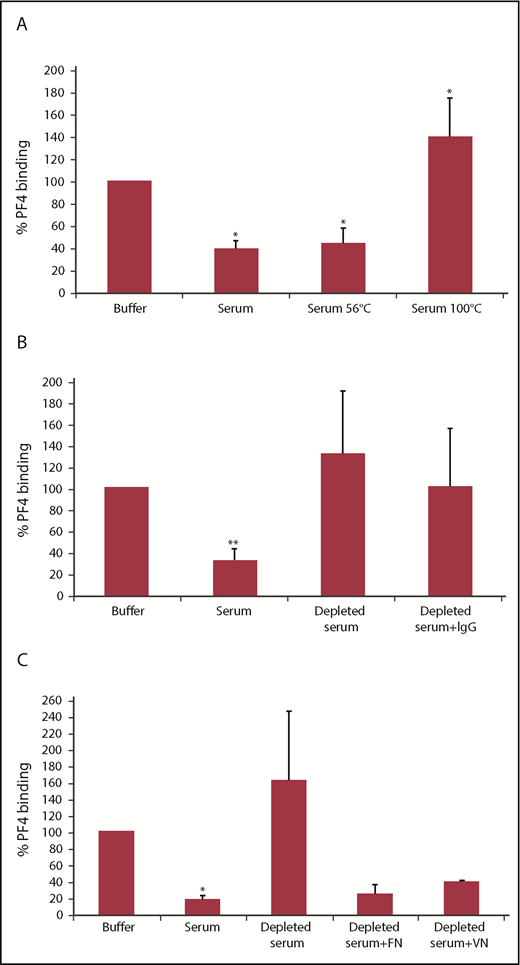
The PF4 binding inhibitor is heat sensitive and binds to Sepharose. (A) GFPs were incubated with 25 µg/mL PF4 in the presence of 20% untreated or heat-treated (56°C or 100°C) serum. Data are mean ± SD of 3 independent experiments. (B) GFPs were incubated with 25 µg/mL PF4 in the presence of 20% untreated serum, IgG-depleted serum (using protein G Sepharose), or IgG-depleted serum substituted with purified IgG according to the original IgG serum concentration. Data are mean ± SD of 10 independent experiments. (C) GFPs were incubated with 25 µg/mL PF4 in the presence of 20% untreated serum, depleted serum (using heparin Sepharose), or depleted serum substituted with 100 µg/mL fibronectin (FN) or 50 µg/mL vitronectin (VN). Data are mean ± SD of 3 (depleted serum + FN or VN, n = 2) independent experiments. (A-C) PF4 binding was detected with an FITC-labeled anti-human PF4 antibody using flow cytometry, and antibody binding was quantified by GMFI. Surface-bound PF4 in the presence of buffer was defined as 100%. *P <.05, **P < .01 vs buffer.
The PF4 binding inhibitor is heat sensitive and binds to Sepharose. (A) GFPs were incubated with 25 µg/mL PF4 in the presence of 20% untreated or heat-treated (56°C or 100°C) serum. Data are mean ± SD of 3 independent experiments. (B) GFPs were incubated with 25 µg/mL PF4 in the presence of 20% untreated serum, IgG-depleted serum (using protein G Sepharose), or IgG-depleted serum substituted with purified IgG according to the original IgG serum concentration. Data are mean ± SD of 10 independent experiments. (C) GFPs were incubated with 25 µg/mL PF4 in the presence of 20% untreated serum, depleted serum (using heparin Sepharose), or depleted serum substituted with 100 µg/mL fibronectin (FN) or 50 µg/mL vitronectin (VN). Data are mean ± SD of 3 (depleted serum + FN or VN, n = 2) independent experiments. (A-C) PF4 binding was detected with an FITC-labeled anti-human PF4 antibody using flow cytometry, and antibody binding was quantified by GMFI. Surface-bound PF4 in the presence of buffer was defined as 100%. *P <.05, **P < .01 vs buffer.
To further test the potential role of α-2-macroglobulin, vitronectin, and fibronectin, we analyzed PF4 binding to GFPs in the presence of each factor separately. Whereas α-2-macroglobulin only had a negligible effect on PF4 binding to GFPs (P >.05 vs buffer; Figure 3A), vitronectin showed significant inhibition starting at a concentration of 3 µg/mL (P = .0230 vs buffer; Figure 3B), and fibronectin reduced PF4 binding significantly beginning at a concentration of 50 µg/mL (P = .0233 vs buffer; Figure 3C). Thus, vitronectin and fibronectin concentrations found in plasma (250-400 µg/mL)29,30 are sufficient to mediate the inhibitory effect. The increase in PF4 binding at lower fibronectin concentrations was due to the fibronectin solvent buffer (Figure 3C). The solvent buffer was not responsible for the inhibitory effect at higher concentrations.
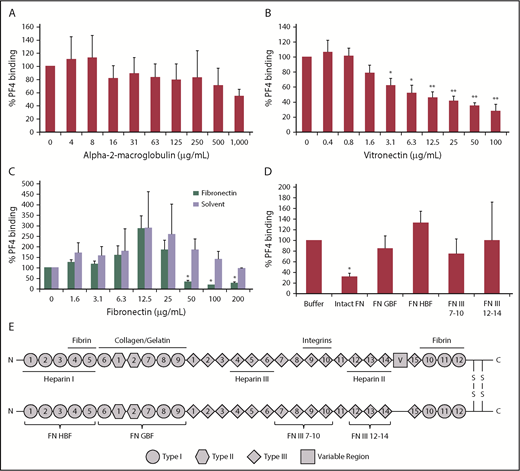
Fibronectin and vitronectin, but not α-2-macroglobulin, inhibit PF4 binding to platelets. GFPs were incubated with 10 µg/mL PF4 in the presence of increasing concentrations of α-2-macroglobulin (A), vitronectin (B), or fibronectin or the respective volumes of its solvent buffer (C). (D) GFPs were incubated with 10 µg/mL PF4 in the presence of 50 µg/mL intact fibronectin (FN), fibronectin gelatin-binding fragment (FN GBF), fibronectin heparin-binding fragment (FN HBF), fibronectin fragments 7-10 of type III domain (FN III 7-10), or fibronectin fragments 12-14 of type III domain (FN III 12-14). (A-D) PF4 binding was detected with an FITC-labeled anti-human PF4 antibody using flow cytometry, and antibody binding was quantified by GMFI. Surface-bound PF4 in the presence of buffer was defined as 100%. Data are mean ± SD of 3 independent experiments. (E) Schematic structure of plasma fibronectin. Fibronectin exists as a dimer, with each monomer composed of 12 type I repeats (circles), 2 type II repeats (hexagons), and 15 type III repeats (diamonds). In contrast to the cellular form, plasma fibronectin lacks the alternatively spliced domains EIIIA and EIIIB (not shown) but includes a variable region (square) in 1 of the subunits. The subunits are linked by 2 disulfide bridges located close to the C termini. Soluble plasma fibronectin occurs as a compact curled-up form and becomes elongated when binding to integrins on cell surfaces. Additional binding sites are indicated for heparin, collagen, gelatin, and fibrin. Brackets denote the fragments used in this study. *P < .05, **P < .001 vs buffer.
Fibronectin and vitronectin, but not α-2-macroglobulin, inhibit PF4 binding to platelets. GFPs were incubated with 10 µg/mL PF4 in the presence of increasing concentrations of α-2-macroglobulin (A), vitronectin (B), or fibronectin or the respective volumes of its solvent buffer (C). (D) GFPs were incubated with 10 µg/mL PF4 in the presence of 50 µg/mL intact fibronectin (FN), fibronectin gelatin-binding fragment (FN GBF), fibronectin heparin-binding fragment (FN HBF), fibronectin fragments 7-10 of type III domain (FN III 7-10), or fibronectin fragments 12-14 of type III domain (FN III 12-14). (A-D) PF4 binding was detected with an FITC-labeled anti-human PF4 antibody using flow cytometry, and antibody binding was quantified by GMFI. Surface-bound PF4 in the presence of buffer was defined as 100%. Data are mean ± SD of 3 independent experiments. (E) Schematic structure of plasma fibronectin. Fibronectin exists as a dimer, with each monomer composed of 12 type I repeats (circles), 2 type II repeats (hexagons), and 15 type III repeats (diamonds). In contrast to the cellular form, plasma fibronectin lacks the alternatively spliced domains EIIIA and EIIIB (not shown) but includes a variable region (square) in 1 of the subunits. The subunits are linked by 2 disulfide bridges located close to the C termini. Soluble plasma fibronectin occurs as a compact curled-up form and becomes elongated when binding to integrins on cell surfaces. Additional binding sites are indicated for heparin, collagen, gelatin, and fibrin. Brackets denote the fragments used in this study. *P < .05, **P < .001 vs buffer.
The inhibitory effect of fibronectin seems to require the intact fibronectin molecule. The gelatin-binding fragment of heparin, the heparin-binding fragment of fibronectin, and those consisting of fragments 7 to 10 or fragments 12 to 14 of the type III domain of fibronectin did not show significant influence (P > .05 vs buffer; Figure 3D). The schematic structure of plasma fibronectin (intact fibronectin) and the fragments used are provided in Figure 3E.
Influence of fibronectin and vitronectin on PF4/heparin complex binding to platelets
Fibronectin (50 µg/mL) (P = .0009; Figure 4A) strongly reduced PF4/heparin complex binding to platelets at concentrations that also inhibited PF4 binding to platelets. The inhibitory activity of vitronectin was less pronounced (P = .3057; Figure 4B).
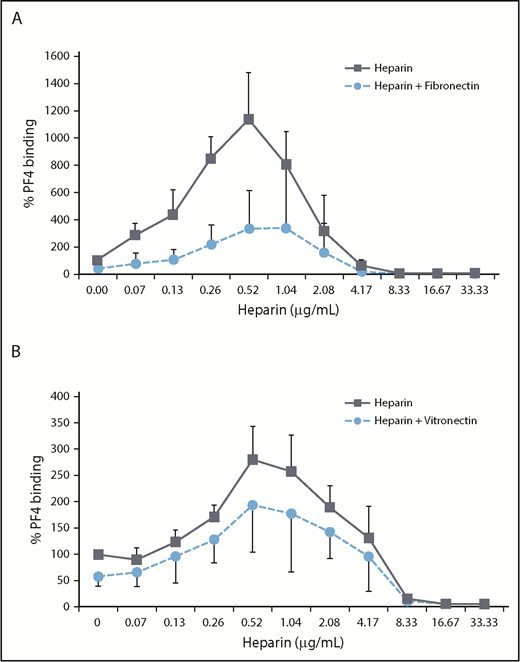
Fibronectin more than vitronectin inhibits PF4/heparin complex binding to platelets. GFPs were incubated with 25 µg/mL PF4 in the presence of increasing concentrations of UFH and 50 µg/mL fibronectin or buffer (A) or UFH and 10 µg/mL vitronectin or buffer (B). PF4 binding was detected with an FITC-labeled anti-human PF4 antibody using flow cytometry, and antibody binding was quantified by GMFI. PF4 binding in the presence of buffer was defined as 100%. Data are mean ± standard deviation of 3 independent experiments.
Fibronectin more than vitronectin inhibits PF4/heparin complex binding to platelets. GFPs were incubated with 25 µg/mL PF4 in the presence of increasing concentrations of UFH and 50 µg/mL fibronectin or buffer (A) or UFH and 10 µg/mL vitronectin or buffer (B). PF4 binding was detected with an FITC-labeled anti-human PF4 antibody using flow cytometry, and antibody binding was quantified by GMFI. PF4 binding in the presence of buffer was defined as 100%. Data are mean ± standard deviation of 3 independent experiments.
Influence of fibronectin and vitronectin on antibody binding to PF4/heparin complexes
Fibronectin reduced anti-PF4/heparin antibody binding to PF4/heparin complexes coated onto a solid phase beginning at a concentration of 100 µg/mL (P = .0173 vs buffer) and was even more pronounced at concentrations present in plasma: 200 µg/mL (P = .0030 vs buffer) and 400 µg/mL (P = .0005 vs buffer). Vitronectin had no effect on anti-PF4/heparin antibody binding. Inhibition with high heparin concentrations served as control (Figure 5A).
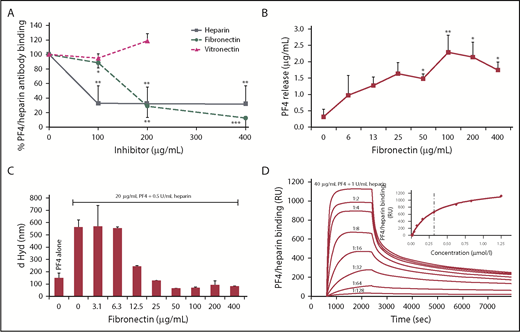
Fibronectin reduces anti-PF4/heparin antibody binding to PF4/heparin complexes by disrupting the complexes. (A) Sera from patients with anti-PF4/heparin IgG were tested in an in-house PF4/heparin ELISA in the presence of increasing concentrations of fibronectin, vitronectin, or heparin, which served as positive control. The signal of anti-PF4/heparin antibody binding without inhibitor was set as 100%. Data are mean ± SD of 5 (vitronectin = 3) independent experiments. (B) The amount of PF4 released from PF4/heparin complexes coated to the microtiter plate by fibronectin was determined by a commercially available ELISA kit. Data are mean ± SD of 3 independent experiments. *P < .05, **P < .01 vs released PF4 without fibronectin. (C) Size of 20 µg/mL PF4 alone and with 0.5 IU/mL UFH plus increasing concentrations of fibronectin was determined by PCS. Data (mean ± SD) represent the results of a minimum of 10 measurements per concentration of fibronectin. One representative experiment is shown. (D) Binding of PF4/heparin complexes (40 µg/mL PF4 + 1 IU/mL UFH) and serial 1:2 dilutions to immobilized fibronectin was monitored by SPR over 7000 seconds. Inset: Half-maximal binding of serial dilutions determines the KD value (dashed line). One representative experiment is shown. *P < .05, **P < .01, ***P < .001 vs antibody binding without inhibitor. d Hyd, hydrodynamic diameter; RU, resonance units.
Fibronectin reduces anti-PF4/heparin antibody binding to PF4/heparin complexes by disrupting the complexes. (A) Sera from patients with anti-PF4/heparin IgG were tested in an in-house PF4/heparin ELISA in the presence of increasing concentrations of fibronectin, vitronectin, or heparin, which served as positive control. The signal of anti-PF4/heparin antibody binding without inhibitor was set as 100%. Data are mean ± SD of 5 (vitronectin = 3) independent experiments. (B) The amount of PF4 released from PF4/heparin complexes coated to the microtiter plate by fibronectin was determined by a commercially available ELISA kit. Data are mean ± SD of 3 independent experiments. *P < .05, **P < .01 vs released PF4 without fibronectin. (C) Size of 20 µg/mL PF4 alone and with 0.5 IU/mL UFH plus increasing concentrations of fibronectin was determined by PCS. Data (mean ± SD) represent the results of a minimum of 10 measurements per concentration of fibronectin. One representative experiment is shown. (D) Binding of PF4/heparin complexes (40 µg/mL PF4 + 1 IU/mL UFH) and serial 1:2 dilutions to immobilized fibronectin was monitored by SPR over 7000 seconds. Inset: Half-maximal binding of serial dilutions determines the KD value (dashed line). One representative experiment is shown. *P < .05, **P < .01, ***P < .001 vs antibody binding without inhibitor. d Hyd, hydrodynamic diameter; RU, resonance units.
Interaction of fibronectin with PF4/heparin complexes
Next, we tested whether fibronectin releases PF4 from PF4/heparin complexes linked to solid phase. The amount of PF4 in the supernatant after incubating the PF4/heparin-coated plates with fibronectin increased analogously to the fibronectin concentration used and reached statistical significance at 13 µg/mL (P = .0276 vs buffer; Figure 5B). The release of PF4 was caused by disruption of PF4/heparin complexes by fibronectin, because addition of fibronectin reduced the size of PF4/heparin complexes, as shown in PCS analysis (Figure 5C). Our SPR studies confirmed a direct interaction of fibronectin with PF4/heparin complexes (Figure 5D).
Influence of fibronectin on anti-PF4/heparin antibody–induced platelet activation
All donor platelets were activated in the presence of 0.2 aFXaU/mL reviparin and anti-PF4/heparin antibodies without fibronectin (lag-time, 16.3 ± 9.4 minutes [mean ± SD]; Figure 6). Addition of fibronectin inhibited activation of platelets at a concentration of 100 µg/mL (inhibition in 12/16 donors; lag-time, 37.8 ± 13.3 minutes [mean ± SD]) and completely abrogated platelet activation at 200 µg/mL. The inhibitory effect of fibronectin was not mediated by competitive binding of fibronectin to the platelet surface. Platelets incubated with fibronectin (200 µg/mL) and washed afterward were still activated by anti-PF4/heparin antibodies (7/7 donors), which further supports that the inhibition of platelet activation by anti-PF4/heparin antibodies is more likely due to PF4/heparin complex disruption by fibronectin in the fluid phase.
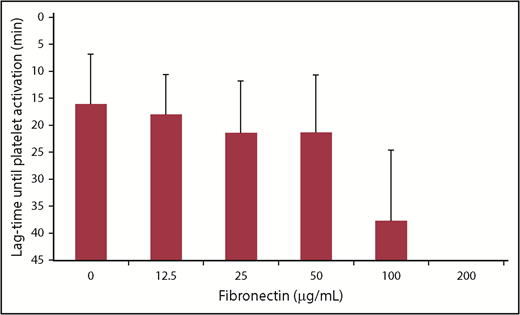
Fibronectin inhibits platelet activation in the HIPA test. Washed platelets were incubated with reviparin (0.2 aFXaU/mL) and anti-PF4/heparin positive sera in the absence or presence of increasing concentrations of fibronectin. Lag-time of platelet activation was monitored for 45 minutes. No observation of platelet activation within 45 minutes was recorded as 45 minutes. Data are mean ± SD of 4 sera tested with 4 platelet donors each (n = 16 donors in total).
Fibronectin inhibits platelet activation in the HIPA test. Washed platelets were incubated with reviparin (0.2 aFXaU/mL) and anti-PF4/heparin positive sera in the absence or presence of increasing concentrations of fibronectin. Lag-time of platelet activation was monitored for 45 minutes. No observation of platelet activation within 45 minutes was recorded as 45 minutes. Data are mean ± SD of 4 sera tested with 4 platelet donors each (n = 16 donors in total).
Fibronectin and anti-PF4/heparin IgG levels in HIT patients and controls
From 2 McMaster University serosurveillance studies linked to prospective clinical trials, we compared the plasma levels of intact fibronectin and of anti-PF4/heparin IgG consisting of 3 types of 59 ELISA+ patients (n = 17 SRA+/ELISA+ patients with clinical HIT; n = 18 SRA+/ELISA+ patients who did not develop clinical HIT; and n = 24 patients who were SRA− but ELISA+) and 30 patients who were SRA−/ELISA−. Intact plasma fibronectin levels were higher in ELISA− patients (median, 109.1 µg/mL; range, 33.8-312.7) compared with all ELISA+ patients (median, 73.8 µg/mL; range, 15.8-262.0; P < .001; Figure 7A). This also applied to the fibronectin levels of ELISA− patients compared with the single groups (1) vs ELISA+/SRA+/clinical HIT (median, 61.3 µg/mL, range, 26.1-99.5; P = .001), (2) vs ELISA+/SRA+/no clinical HIT (median, 80.6 µg/mL; range, 40.9-219.5; P = .036); and (3) vs ELISA+/SRA−/no clinical HIT (median, 80.3 µg/mL; range, 15.8-262.0 µg/mL; P = .011). When we compared the fibronectin levels over all 4 groups, there was a clear trend of an increase from ELISA+/SRA+/clinical HIT to ELISA-/SRA- (Figure 7A; P = .009 for trend). The fibronectin levels were also nominally higher in ELISA+/SRA+ patients who did not develop clinical HIT (median, 80.6 µg/mL; range, 40.9-219.5) compared with ELISA+/SRA+ patients with clinical breakthrough of HIT (median, 61.3 µg/mL; range, 26.1-99.5; P = .062).
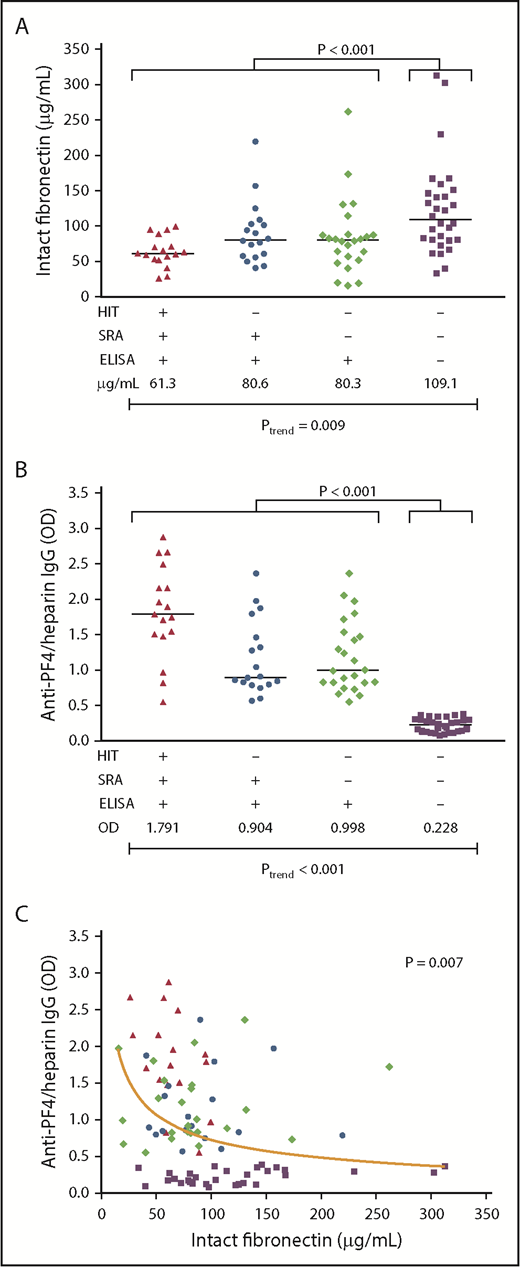
Fibronectin plasma levels correlate with HIT antibody formation. Plasma levels of intact fibronectin (in micrograms per milliliter) (A) and of anti-PF4/heparin IgG (OD) (B) in patients with clinically confirmed HIT who showed a positive SRA and a positive PF4/heparin IgG ELISA (▲, n = 17); patients with no clinical HIT, but a positive SRA and ELISA (●, n = 18); patients with no clinical HIT, a negative SRA, and a positive ELISA (♦, n = 24); and patients who did not develop anti-PF4/heparin IgG (▪, n = 30). Data are intact fibronectin and anti-PF4/heparin IgG levels of individual plasma and median of each group. The P values are provided for the comparison between levels of all ELISA-positive and ELISA-negative patients and Ptrend over all 4 groups. (C) Intact fibronectin levels (in micrograms per milliliter) are plotted against anti-PF4/heparin IgG OD levels of individual patient plasmas, including all 4 groups (n = 89). A nonlinear association between fibronectin and anti-PF4/heparin IgG plasma levels is illustrated by the nonlinear regression curve, as determined by median regression. Regression formula: y = 0.102 + 0.825*1/SQRT(x/100).
Fibronectin plasma levels correlate with HIT antibody formation. Plasma levels of intact fibronectin (in micrograms per milliliter) (A) and of anti-PF4/heparin IgG (OD) (B) in patients with clinically confirmed HIT who showed a positive SRA and a positive PF4/heparin IgG ELISA (▲, n = 17); patients with no clinical HIT, but a positive SRA and ELISA (●, n = 18); patients with no clinical HIT, a negative SRA, and a positive ELISA (♦, n = 24); and patients who did not develop anti-PF4/heparin IgG (▪, n = 30). Data are intact fibronectin and anti-PF4/heparin IgG levels of individual plasma and median of each group. The P values are provided for the comparison between levels of all ELISA-positive and ELISA-negative patients and Ptrend over all 4 groups. (C) Intact fibronectin levels (in micrograms per milliliter) are plotted against anti-PF4/heparin IgG OD levels of individual patient plasmas, including all 4 groups (n = 89). A nonlinear association between fibronectin and anti-PF4/heparin IgG plasma levels is illustrated by the nonlinear regression curve, as determined by median regression. Regression formula: y = 0.102 + 0.825*1/SQRT(x/100).
Corresponding anti-PF4/heparin IgG levels were ELISA− patients (median optical density [OD], 0.228; range, 0.082-0.385) compared with the single groups (1) vs ELISA+/SRA+/clinical HIT (median OD, 1.791; range, 0.55-2.875; P < .001), (2) vs ELISA+/SRA+/no clinical HIT (median OD, 0.904; range, 0.569-2.362; P < .001), (3) vs ELISA+/SRA−/no clinical HIT (median OD, 0.998; range, 0.554-2.362; P < .001). Over all 4 groups, P < .001 for trend (Figure 7B).
Finally, we found a nonlinear association between intact fibronectin and anti-PF4/heparin IgG plasma levels, including samples from all 4 groups (P = .007; Figure 7C).
Discussion
Our study identifies fibronectin as interfering with formation of PF4/heparin complexes. This classifies fibronectin levels as a potential risk factor for HIT, likely acting via 2 mechanisms. First, higher levels of fibronectin are associated with a lower risk for immunization against PF4/heparin complexes. Second, fibronectin might also modulate the risk of clinical breakthrough of HIT among immunized patients who have already formed platelet-activating antibodies.
All prospective studies of seroconversion of anti-PF4/heparin antibodies show that different patient populations differ widely in their immune response toward PF4/heparin complexes. Also, those patients sharing anti-PF4/heparin antibodies with similar in vitro characteristics can differ considerably in their clinical breakthrough of HIT, ranging from being completely asymptomatic, to a moderate decrease in platelet counts, to the typical picture of severe HIT with a >50% decrease in platelet counts and development of new thrombotic complications.14-16,31,32
Several risk factors have been identified that appear to modify the risk for HIT. UFH vs LMWH15 and major vs minor surgery33 appear to be the most significant factors. Also, genetic risk factors associated with an enhanced risk for HIT, such as polymorphisms of the ACP1 gene34 or the interleukin 10 promoter,35 are associated with differences in antibody levels. More directly related to platelet activation by anti-PF4/heparin antibodies is the number of FcγRIIAs on the platelet surface,36 polymorphisms determining signal transduction by the FcγRIIA,37 or the interaction of FcγRIIA with IgG, such as the R131H polymorphism.38,39 This last characteristic increases breakthrough of thrombosis, in the case of FcγRIIA R131 homozygosity.
We first examined factors present in both serum and plasma, particularly the role of IgG. This approach was based on the well-known observation that anti-PF4/heparin antibodies activate washed platelets more strongly compared with platelets within PRP obtained from the same donor.17 In our study, serum and plasma were potent in inhibiting PF4 binding to platelets. Earlier findings22 show that IgG depletion of the serum enhanced sensitivity of the assay; however, to our surprise, reconstitution of the serum with the depleted IgG fraction did not restore the inhibitory effect. Therefore, we assessed binding of proteins other than IgG to protein G Sepharose by mass spectrometry and found several potential candidates; 2 of which, fibronectin and vitronectin, interfered with PF4 binding to platelets. However, only fibronectin inhibited binding of PF4/heparin complexes to platelets and binding of anti-PF4/heparin antibodies to immobilized PF4/heparin complexes. Furthermore, fibronectin directly binds to PF4/heparin complexes, as shown by SPR, leading to disruption of PF4/heparin complexes in the surface-bound and soluble phase, as shown by measuring the release of PF4 from PF4/heparin-coated ELISA plates and the reduction in complex size by PCS. When we aimed to characterize the parts of fibronectin responsible for interference with PF4 binding, none of the fibronectin fragments had any significant effect. This suggests that intact fibronectin is required for inhibiting PF4 binding to platelets and might also account for the interaction with PF4/heparin complexes. Whereas fibronectin binding to several integrins on cell surfaces40 might interfere with PF4 binding to platelets, the inhibition of platelet activation by anti-PF4/heparin antibodies is likely due to direct interaction with PF4/heparin complexes. Because fibronectin has 3 distinct binding domains for heparin (Figure 3E), it is intriguing to assume that fibronectin interferes with PF4/heparin complexes by binding to heparin; however, we cannot exclude that fibronectin also directly interacts with PF4.
Our observations provide a potential explanation for why assays using washed platelets are more sensitive for detecting platelet-activating antibodies compared with PRP-based or whole-blood assays. In addition, they help to explain the recent observation that addition of PF4 to platelets increases the sensitivity of functional HIT tests.41-43 By increasing the concentration of PF4, binding of fibronectin is likely outcompeted.
These in vitro findings led to the intriguing hypothesis that fibronectin might also be a risk factor for HIT. To address this hypothesis, we took advantage of blood samples from 2 prospective serosurveillance studies of heparin-treated patients. This allowed us to compare the plasma fibronectin levels in patients with different types of immune reaction against PF4/heparin and to correlate this with the clinical breakthrough of HIT. Consistent with our in vitro studies and with our hypothesis, we found lower plasma fibronectin levels in patients who developed anti-PF4/heparin antibodies compared with patients who did not. In addition, those patients who tested positive in the ELISA and the SRA had a >50% decrease in platelet count, indicating that a diagnosis of HIT was associated with lower fibronectin levels compared with patients who showed a similar in vitro antibody pattern (ie, SRA+/ELISA+) but did not develop clinical HIT (either a platelet count decrease or a thrombosis). However, the difference between both groups was not demonstrative (P = .062) and is not conclusive to clearly associate low fibronectin levels with clinical breakthrough. However, we found a highly significant trend for increasing levels of fibronectin over the following 4 patient groups, classified from the highest to lowest expression of HIT and HIT-associated immunization: SRA+/ELISA+/clinical HIT < SRA+/ELISA+/no HIT ∼ SRA−/ELISA+/no HIT < SRA−/ELISA−/no HIT (Ptrend = .009). The overlap between groups might result from the modulating effect of PF4 levels44 (and, consequently, PF4/heparin complexes), which likely vary among patients and likewise influence PF4/heparin immunization and breakthrough of HIT.
It is very challenging to identify patients with a positive PF4/heparin ELISA and a positive functional assay (HIPA or SRA) who do not have HIT, because patients are usually thrombocytopenic, which triggers diagnostic testing for HIT antibodies. Thus, identifying true ELISA+/SRA+/HIT− patients requires large serosurveillance studies. To the best of our knowledge, there is no other study available in which a sufficient number of patients developed HIT to make a meaningful comparison of fibronectin levels in patients who are SRA+ and ELISA+ and had clinical breakthrough of HIT and patients who are SRA+ and ELISA+ but had no clinical breakthrough of HIT. In addition, stability of any measured protein is an issue for comparative studies. The major advantage of the McMaster University study is that samples were obtained at the same time and stored under the same conditions. The limitation of our study is that we found a significant trend over all groups but not a clear difference in fibronectin levels between single groups. In conclusion, fibronectin likely modifies the risk of HIT but is certainly not the only cause for the development of HIT.
There are some intriguing implications that arise from our studies. It is known that patients with surgery are more likely to develop HIT compared with medical patients45 and that patients with major trauma are more likely to develop HIT compared with patients with minor trauma.33 There is a characteristic early decrease in platelet count that reflects hemodilution (related to fluid administration) and platelet activation/consumption (related to perioperative hemostasis). It is possible that the combination of hemodilution and platelet activation in surgical patients results in an early postoperative decrease in fibronectin (hemodilution and consumption)46-48 and increase in PF4 (platelet activation), conditions that would favor formation of potentially immunizing PF4/heparin complexes vs medical patients who are treated with heparin.
In summary, we show that fibronectin interferes with the generation of PF4/heparin complexes, with binding of PF4/heparin complexes to platelets, and also inhibits platelet activation by anti-PF4/heparin antibodies. Furthermore, low fibronectin levels are associated with a higher immunization rate for PF4/heparin complexes and might also be a risk factor for clinical breakthrough of HIT if platelet-activating anti-PF4/heparin antibodies are present.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Till Ittermann (Universität Greifswald) for help with the statistical analysis of the clinical studies.
This work was supported by a grant from the Bundesministerium für Bildung und Forschung (FKZ 03Z2CN11 and FKZ 03Z2CN12) and was funded by the Deutsche Forschungsgemeinschaft (German Research Foundation) Graduiertenkolleg 840 and Projektnumber 374031971-TRR 240 (S.H. and A.G.).
Authorship
Contribution: K.K., A.G., T.E.W., and S.H. designed the research; K.K., P.P., C.T., S.B., I.J., M.M., and E.H. performed experiments and analyzed the data; and K.K., A.G., and T.E.W. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Andreas Greinacher, Institut für Immunologie und Transfusionsmedizin, Universitätsmedizin Greifswald, Ferdinand-Sauerbruch-Str, D-17489 Greifswald, Germany; e-mail: greinach@unigreifswald.de.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal