

Key Points
Small-molecule–induced BTK degradation has antiproliferative effects superior to inhibition alone in cancer cells.
The lead degrader DD-03-171 reduces tumor burden and extends survival in lymphoma patient-derived xenograft models.

Abstract
The covalent Bruton tyrosine kinase (BTK) inhibitor ibrutinib is highly efficacious against multiple B-cell malignancies. However, it is not selective for BTK, and multiple mechanisms of resistance, including the C481S-BTK mutation, can compromise its efficacy. We hypothesized that small-molecule–induced BTK degradation may overcome some of the limitations of traditional enzymatic inhibitors. Here, we demonstrate that BTK degradation results in potent suppression of signaling and proliferation in cancer cells and that BTK degraders efficiently degrade C481S-BTK. Moreover, we discovered DD-03-171, an optimized lead compound that exhibits enhanced antiproliferative effects on mantle cell lymphoma (MCL) cells in vitro by degrading BTK, IKFZ1, and IKFZ3 as well as efficacy against patient-derived xenografts in vivo. Thus, “triple degradation” may be an effective therapeutic approach for treating MCL and overcoming ibrutinib resistance, thereby addressing a major unmet need in the treatment of MCL and other B-cell lymphomas.
Introduction
Bruton tyrosine kinase (BTK) is a TEC-family nonreceptor tyrosine kinase that signals downstream of numerous cellular receptors, including the B-cell receptor (BCR), toll-like receptors, and Fc receptors.1 BTK plays a particularly important role in B-cell development and function and is critical for progression into the cell cycle and proper B-cell activation,2-4 and loss-of-function mutations in BTK result in X-linked agammaglobulinemia due to a severe defect in B-cell development.5 Importantly, BTK transduces constitutive signaling downstream of the BCR in many B-cell malignancies, so BTK has long been considered an attractive target for treating these diseases. Indeed, the clinically approved covalent BTK inhibitor ibrutinib has been approved for use in patients with mantle cell lymphoma (MCL), chronic lymphocytic leukemia, Waldenström macroglobulinemia, and marginal zone lymphoma.6
Despite ibrutinib’s success in these indications, both intrinsic and acquired resistance has been observed in the clinic. For example, roughly one-third of patients with MCL fail to respond to ibrutinib (intrinsic resistance), which may be due in part to activation of nonclassical NF-κB signaling.7 For those who do respond, acquired resistance quickly arises, often due to the C481S mutation of BTK, which prevents ibrutinib from forming a covalent bond with BTK and significantly reduces its potency, resulting in median progression-free survival of only ∼14 months.8 In fact, a “real-world” report of ibrutinib use in patients with MCL suggested that median time to progression or drug cessation due to toxicity is only 8 months.9 Thus, the development of therapeutic strategies capable of preventing or overcoming BTK inhibitor resistance is an urgent unmet medical need for patients with MCL and other B-cell malignancies.
Previously, we reported that HSP90 inhibition induced almost complete loss of BTK and other client proteins.10 Correspondingly, HSP90 inhibition, through its effect on both BCR and nonclassical NF-κB signaling, reduced the viability of ibrutinib-sensitive and ibrutinib-resistant MCL cell lines, both in vitro and in patient-derived xenograft (PDX) models in vivo. However, to date, there are no clinically approved HSP90 inhibitors, as most trials have been associated with limited efficacy and significant toxicity, presumably because HSP90 inhibition promotes the degradation of different substrates in different tissues and rapidly induces stress responses that may mediate resistance.11
Thus, we turned to a small-molecule–mediated protein degradation platform that we and others have recently pioneered.12-15 Small-molecule degraders,16 also referred to as proteolysis-targeting chimeras or degronimids,17 contain an E3 ligase-targeting moiety connected via a linker to a ligand for a target of interest. Degraders bring an endogenous E3 ligase into close proximity with the target, leading to its ubiquitination and subsequent proteasomal degradation. Small-molecule–induced degradation of proteins is an emerging pharmacological strategy that holds significant therapeutic promise,18 but not all ligandable targets are readily degradable. To determine which members of the kinome are amenable to this mode of pharmacological targeting, we previously generated a degrader that utilizes a promiscuous, multitargeted kinase inhibitor as its warhead.14 While this compound efficiently bound to a large subset of the kinome, proteomic analysis revealed that not all kinases bound were effectively degraded. Notably, BTK scored as one of the most degraded kinases, indicating that it is a tractable target.
Some of the most commonly employed E3 ligase ligands are thalidomide and its derivatives, lenalidomide and pomalidomide, commonly referred to as IMiDs (immunomodulatory imide drugs). These agents are small-molecule ligands of cereblon (CRBN),19 a substrate adaptor for the ubiquitously expressed cullin ring ligase 4 (CUL4)-RBX1-DDB1-CRBN (CUL4CRBN) E3 ligase. Interestingly, thalidomide interacts with CRBN to form a novel surface, resulting in interactions with neosubstrates such as Ikaros (IKZF1) and Aiolos (IKZF3) and their ubiquitination and subsequent proteasomal degradation.20,21 This activity alone has potent antitumor effects in some liquid malignancies, and lenalidomide (Revlimid) is US Food and Drug Administration approved for the treatment of MCL, multiple myeloma, and myelodysplastic syndromes with deletion of chromosome 5q. Lenalidomide is also undergoing late-stage clinical trials for a number of lymphomas, including MCL and the activated B-cell subtype of diffuse large B-cell lymphoma (ABC DLBCL).
Here, we investigated the utility of small-molecule imide-based degraders of BTK. Specifically, we describe highly potent and selective BTK degraders with efficacy in cellular models of lymphoma and leukemia that can circumvent ibrutinib resistance. Moreover, we discovered that chemical structure modifications enabled us to tune IKZF1/3 degradation activity, allowing us to synthesize a degrader molecule that combined IKZF1/3 degradation with BTK degradation. Importantly, this “triple degrader” had enhanced potency against B-cell malignancies and significant efficacy in PDX models in vivo.
Methods
In vitro CRBN-BTK dimerization
BODIPY-labeled CRBN in complex with damage-specific DNA-binding protein 1 (DDB1) harboring an internal deletion of the flexible β propeller B domain (DDB1ΔB-CRBN)22 and in vitro–biotinylated BTK were treated with increasing concentrations of the indicated compounds in the presence of tracer amounts of terbium-streptavidin. Compound-induced recruitment of BTK to CRBN was quantified following the 520/490 time-resolved fluorescence resonance energy transfer (FRET) ratio22 using a Pherastar plate reader (BMG) utilizing 2 synchronized photomultiplier tubes to reduce background.
Cellular CRBN engagement
Cellular CRBN engagement was determined by interrogating the ability of BTK degraders or lenalidomide as control to outcompete binding of the BRD4 degrader dBET6 to CRBN. Cells stably expressing a BRD4BD2-GFP fusion protein and mCherry from the same messenger RNA (separated by a P2A splice site)22 were treated with dBET6 at 100 nM and increasing concentrations of BTK degraders or lenalidomide as a control. The GFP/RFP signal ratio was quantified using an Acumen laser scanning cytometer (TTP Labtech). Compounds capable of penetrating the cell membrane and binding to CRBN precluded dBET6 from inducing degradation of BRD4BD2-GFP, resulting in a dose-dependent increase in the GFP signal.
Cell lines
Mino, Granta-519, and Maver-1 cells were cultured in RPMI-1640 media supplemented with 10% to 20% fetal bovine serum and 1% penicillin/streptomycin in a 37°C incubator with 5% CO2. TMD8 and HBL1 cells were cultured in IMDM media supplemented with 10% fetal bovine serum (Sigma) and 1% penicillin/streptomycin (Thermo Fisher Scientific) in a 37°C incubator with 5% CO2.
C481S-BTK–expressing cell models
Cells that overexpress wild-type (WT) or BTK with the Cys481Ser mutation (C481S-BTK) were prepared as described previously.23 Briefly, TMD8 or HBL1 cells were transduced with a lentiviral expression vector (pLVX-EF1α-IRES-Puro vector; Clontech Laboratories), with coding sequences for WT- or C481S-BTK. Following lentiviral transduction, stable cell lines were selected by 0.5∼1.0 µg/mL puromycin. Equivalent expression for WT- and C481S-BTK in transduced cells was confirmed by immunoblots.
Western blots and antibodies
Cells were lysed in M-PER buffer (Thermo Scientific) containing protease/phosphatase inhibitor cocktail (Roche). Protein concentrations were measured using a BCA assay (Pierce). Equivalent amounts of protein for each sample were loaded on 4% to 12% Bis-Tris gels (Invitrogen), transferred to nitrocellulose membranes (Bio-Rad), and immunoblotted with antibodies against BTK, hematopoietic cell kinase (HCK), phospho-Erk1/2 (Thr202/Tyr204), Erk1/2, phospho-S536-p65, p65, IKZF1, IKZF3, and actin (Cell Signaling). IRDye800-labeled goat anti-rabbit immunoglobulin G and IRDye680-labeled goat anti-mouse immunoglobulin G (LI-COR) secondary antibodies were used and detected on an Odyssey CLx system.
Proliferation assays
Proliferation assays were performed by treating cells with compounds at the concentrations indicated for 72 hours. Antiproliferative effects of compounds were assessed using CellTiter-Glo luminescent cell viability assays (Promega). The 50% effective dose (ED50) values were calculated with GraphPad Prism nonlinear regression curve fit.
Synergy assays
The CellTiter-Glo assay was used to assess the dose–response of the BTK inhibitor/degrader alone or in combination with other inhibitors. Compounds were dispensed using the JANUS Automated Workstation (PerkinElmer) into 384-well plates preseeded with cells by the MultiFlo FX dispenser (BioTek Instruments). Cells were incubated with compounds for 72 hours at 37°C, and viability was assessed by luminescent measurements using the 2104 Envision Multilabel Reader (PerkinElmer). Synergy was assessed by CalcuSyn 2.0 software (Biosoft, Cambridge, United Kingdom) as previously described.24
Liquid chromatography-mass spectrometry data analysis
Sample preparation and data acquisition for tandem mass tag mass spectrometry was carried out as previously described.25 Proteome Discoverer 2.2 (Thermo Fisher) was used for RAW file processing and controlling peptide and protein level false discovery rates, assembling proteins from peptides, and protein quantification from peptides. Tandem mass spectrometry spectra were searched against a UniProt human database (September 2016) with both the forward and reverse sequences. Database search criteria were as follows: tryptic with 2 missed cleavages, a precursor mass tolerance of 20 ppm, fragment ion mass tolerance of 0.6 Da, static alkylation of cysteine (57.02146 Da), static tandem mass tag (TMT) labeling of lysine residues and N termini of peptides (229.16293 Da), and variable oxidation of methionine (15.99491 Da). TMT reporter ion intensities were measured using a 0.003 Da window around the theoretical m/z for each reporter ion in the MS3 scan. Peptide spectral matches with poor quality MS3 spectra were excluded from quantitation (summed signal-to-noise across 10 channels <200 and precursor isolation specificity <0.5), and resulting data were filtered to only include proteins that had a minimum of 3 unique peptides identified. Reporter ion intensities were normalized and scaled using in-house scripts in the R framework.26 Statistical analysis was carried out using the limma package within the R framework.27
Generation of animal models
PDX models (supplemental Table 5, available on the Blood Web site) were engrafted in Nod.Cg-PrkdcscidIL2rgtm1Wjl/SzJ (NSG) mice purchased from The Jackson Laboratory and handled according to a Dana-Farber Cancer Institute’s Institutional Animal Care and Use Committee–approved protocol (#13-034), as described previously.28 Detailed description of models used and procedures can be found in supplemental Methods and at www.PRoXe.org.
See supplemental Methods for a description of protein docking, mass spectrometry, and chemical synthesis methods, and supplemental Table 5 for PDX model characterization.
Results
Biochemical and mechanistic characterization of BTK degraders
To develop efficient degraders of BTK, we designed compounds based off of the previously reported selective BTK inhibitor CGI174629 and synthesized BTK degraders with polyethylene glycol (DD-03-007) or saturated hydrocarbon chain (DD-03-171) linkers conjugating the parent inhibitor to thalidomide (Figure 1A). As a negative control, we synthesized DD-03-033 (Figure 1A), an analog of DD-03-007 that lacks a carbonyl on the glutarimide ring of the thalidomide moiety, a modification that significantly reduces its affinity for CRBN.14 We also generated DD-04-118 (Figure 1A), an analog which differs from DD-03-171 by only a single atom change of the thalidomide aryl amine nitrogen to an oxygen, resulting in an aryl ether, a change that was predicted to compromise its ability to recruit IKZF1/3 to CRBN.22 Crystal structures of inhibitor-bound BTK guided our selection of the piperazine as a suitable solvent-exposed linker attachment site. This selection was further validated by docking studies identifying the piperazine residue as an appropriate linker attachment site with the correct orientation of linker exit to bridge to an imide ligand bound to CRBN (Figure 1B).
![Figure 1. Structure and biochemical characterization of BTK degraders. (A) Chemical structure and biochemical BTK IC50 of BTK degraders. (B) Docking of lenalidomide-bound CRBN (Protein Data Bank [PDB]: 5FQD; shown as a gray surface) with apo BTK (PDB: 5P9J). BTK is overlaid with CGI1746-bound BTK (PDB: 3OCS; shown in blue). (C) TREEspot visualization of the kinome selectivity profile of DD-03-171 (1 μM). (D) Compound-induced ternary complex formation of recombinant BTK and CRBN-DDB1 (Lanthascreen duplicate mean ± standard error of the mean). (E) Assessment of cellular CRBN engagement by displacement of dBET6 by a degrader. Cells stably expressing BRD4BD2-GFP with mCherry reporter were treated with 100 nM dBET6 and titration of compound. Percentage of GFP/RFP ratio of RFP-positive cells calculated from images using CellProfiler. Data are shown as means ± standard deviation of cell culture duplicates (n = 2). A.U., arbitrary units; IC50, 50% inhibitory concentration.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/133/9/10.1182_blood-2018-07-862953/4/m_blood862953f1.png?Expires=1765898324&Signature=127pEyUd0Yiyok9r78NaUXXdFUCh1hkdA7o5J65TtH1WxuhPL9A~m4iYV~p2Ax85Bj7rWBiQlqPp1kNmzKa-~eS7WqlHsYwEZz31DmzkOUuOPeRq-T6DmQOaTipmGft3rM6C9LOXFnaz~HljLS-G~HnpAKO7Vedat0p3vZT2i6xtmCdkId8AaBXlFKRGDCvZ9bwxP77JGJrpJI7SJB51nA5weZfAG33Y5JA74XH0IacYzkzmAssHyQUv1TmpxpxBAur0YCSBGpPlsA1EZNM9RJcamUeifMeJXPi7AElgQvPFxmG8irxo6ByUiUFIp12v-GGXHDb~vrWIlRkD~J7bLA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Structure and biochemical characterization of BTK degraders. (A) Chemical structure and biochemical BTK IC50 of BTK degraders. (B) Docking of lenalidomide-bound CRBN (Protein Data Bank [PDB]: 5FQD; shown as a gray surface) with apo BTK (PDB: 5P9J). BTK is overlaid with CGI1746-bound BTK (PDB: 3OCS; shown in blue). (C) TREEspot visualization of the kinome selectivity profile of DD-03-171 (1 μM). (D) Compound-induced ternary complex formation of recombinant BTK and CRBN-DDB1 (Lanthascreen duplicate mean ± standard error of the mean). (E) Assessment of cellular CRBN engagement by displacement of dBET6 by a degrader. Cells stably expressing BRD4BD2-GFP with mCherry reporter were treated with 100 nM dBET6 and titration of compound. Percentage of GFP/RFP ratio of RFP-positive cells calculated from images using CellProfiler. Data are shown as means ± standard deviation of cell culture duplicates (n = 2). A.U., arbitrary units; IC50, 50% inhibitory concentration.
Structure and biochemical characterization of BTK degraders. (A) Chemical structure and biochemical BTK IC50 of BTK degraders. (B) Docking of lenalidomide-bound CRBN (Protein Data Bank [PDB]: 5FQD; shown as a gray surface) with apo BTK (PDB: 5P9J). BTK is overlaid with CGI1746-bound BTK (PDB: 3OCS; shown in blue). (C) TREEspot visualization of the kinome selectivity profile of DD-03-171 (1 μM). (D) Compound-induced ternary complex formation of recombinant BTK and CRBN-DDB1 (Lanthascreen duplicate mean ± standard error of the mean). (E) Assessment of cellular CRBN engagement by displacement of dBET6 by a degrader. Cells stably expressing BRD4BD2-GFP with mCherry reporter were treated with 100 nM dBET6 and titration of compound. Percentage of GFP/RFP ratio of RFP-positive cells calculated from images using CellProfiler. Data are shown as means ± standard deviation of cell culture duplicates (n = 2). A.U., arbitrary units; IC50, 50% inhibitory concentration.
To determine whether conjugation of E3 ligase binders to BTK inhibitors would impede the ability of these bivalent molecules to inhibit BTK, we tested these compounds in a commercial FRET-based BTK kinase assay (Invitrogen Z’-Lyte; supplemental Table 1). Importantly, degraders had comparable activity to the parent inhibitor, validating our choice of linker attachment site. We also tested binding of DD-03-171 against a panel of 468 kinases (KINOMEscan) and found that similar to CGI1746, DD-03-171 binds exclusively to BTK at a screening concentration of 1 µM (Figure 1C).
Next, we determined whether our CRBN-binding degraders could induce dimerization of CRBN and BTK. Using a time-resolved FRET-based biochemical assay, we found that DD-03-007, DD-03-171, and DD-04-118 efficiently induced dimerization of BTK and CRBN, while neither lenalidomide nor the CRBN-nonbinding compound DD-03-033 could do so (Figure 1D). The broader peak of DD-03-171 vs DD-03-007 may be because DD-03-171 exhibits greater cooperativity of BTK-degrader-CRBN complex formation.
Lead compounds potently and selectively degrade BTK in a proteasome- and CRBN-dependent manner
Next, we assessed the ability of these degrader compounds to destabilize BTK in Ramos B cells. Both DD-03-007 and DD-03-171 significantly reduced cellular BTK levels at concentrations as low as 100 nM within 4 hours of treatment, while DD-03-033, a negative control that does not bind CRBN, did not (Figure 2A). This suggests that the cellular permeability of these compounds was sufficient to induce BTK degradation. Furthermore, both DD-03-007 and DD-03-171, but not DD-03-033, potently inhibited phosphorylation of Erk1/2, a downstream target of BTK, upon stimulation of the BCR (Figure 2B). To assess the durability of BTK degradation, we treated cells with DD-03-007 or DD-03-171 for 2 hours and collected cells at various time points after compound washout. Consistent with the extended half-life of BTK in primary B cells,31 we found that BTK levels in degrader-treated cells remained below levels in vehicle-treated cells even 24 hours after washout (Figure 2C), illustrating that BTK degraders induce sustained depletion of BTK.
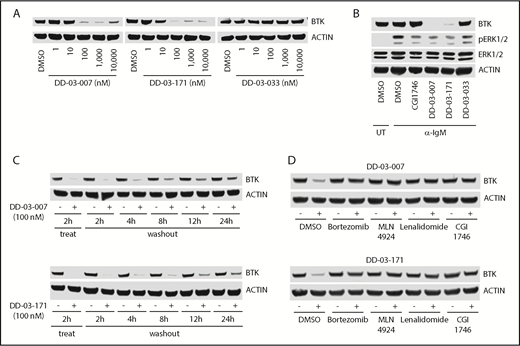
BTK degraders induce potent and sustained BTK degradation dependent on CRBN, neddylation, and the proteasome. (A) Immunoblots from Ramos B cells treated as indicated for 4 hours. (B) Immunoblots from Ramos B cells pretreated with the indicated compounds (1 μM) for 4 hours followed by stimulation with α-immunoglobulin M (10 μg/mL) for 30 minutes. (C) Immunoblots from Ramos B cells treated for 2 hours with indicated compounds, followed by washout for indicated times. (D) Immunoblots from Ramos B cells pretreated for 1 hour with bortezomib (500 nM), MLN4924 (1 μM), lenalidomide (10 μM), or CGI1746 (10 μM) and then treated with either DD-03-007 or DD-03-171 (100 nM) for 4 hours. DMSO, dimethyl sulfoxide; IgM, immunoglobulin M; UT, untreated.
BTK degraders induce potent and sustained BTK degradation dependent on CRBN, neddylation, and the proteasome. (A) Immunoblots from Ramos B cells treated as indicated for 4 hours. (B) Immunoblots from Ramos B cells pretreated with the indicated compounds (1 μM) for 4 hours followed by stimulation with α-immunoglobulin M (10 μg/mL) for 30 minutes. (C) Immunoblots from Ramos B cells treated for 2 hours with indicated compounds, followed by washout for indicated times. (D) Immunoblots from Ramos B cells pretreated for 1 hour with bortezomib (500 nM), MLN4924 (1 μM), lenalidomide (10 μM), or CGI1746 (10 μM) and then treated with either DD-03-007 or DD-03-171 (100 nM) for 4 hours. DMSO, dimethyl sulfoxide; IgM, immunoglobulin M; UT, untreated.
To verify the mechanisms of action of these BTK degraders, we cotreated cells with a proteasome inhibitor (bortezomib) or a NEDD8-activating enzyme E1 inhibitor (MLN-492432 ) and found that both inhibitors prevented degradation of BTK (Figure 2D). Moreover, we performed competition experiments with either lenalidomide (for CRBN) or CGI1746 (for BTK) and found that excess quantities of either parent compounds could also prevent BTK degradation (Figure 2D). Thus, these BTK degraders act in a proteasome- and CRBN-dependent manner.
BTK degradation overcomes ibrutinib resistance and synergizes with HCK inhibition in ABC DLBCL
To explore the pharmacological consequences of BTK degradation in cellular models of cancer, we investigated the potential of our BTK degraders to overcome the increasingly urgent problem of acquired ibrutinib resistance in the clinic. While the most common ibrutinib-resistant BTK mutant is C481S, other mutants observed in patients include C481R, C481F, C481Y, T474I, and T474S.33 New-generation noncovalent BTK inhibitors have been developed that retain activity against the Cys481 and gatekeeper Thr474 point mutants.34 Many of these noncovalent inhibitors have activity on WT and C481-mutant BTK with equal potency, and some even consistently inhibit all mutants. Thus, we hypothesized that degraders based on CGI1746 (an inhibitor with a similar structure and BTK binding mode as these new-generation inhibitors) would be able to degrade clinically relevant mutant forms of BTK.
To assess the ability of our degraders to overcome the C481S-BTK mutation, we treated TMD8 ABC DLBCL cells that expressed WT- or C481S-BTK with DD-03-007 and DD-03-171. Both degraders induced degradation of WT- and C481S-BTK, although DD-03-007 was slightly less potent (Figure 3A). We then compared the antiproliferative effects of CGI1746, ibrutinib, DD-03-007, and DD-03-171 on each cell line. While C481S-BTK expression reduced the potency of ibrutinib ∼1000-fold, it had minimal effects on the potency of DD-03-007 or DD-03-171 (Figure 3B); this was similarly true in HBL1 ABC DLBCL cells (supplemental Figure 1B). Based on previous reports, the potent effects of ibrutinib in BTK-WT TMD8 and HBL1 cells likely resulted from numerous off-target effects on other TEC and SRC family kinases,35-37 which are not recapitulated with the degraders. Taken together, these data illustrate that BTK degradation can overcome a clinically relevant form of acquired ibrutinib resistance.

BTK degraders overcome ibrutinib resistance in ABC DLBCL. (A) Immunoblots of TMD8 cells overexpressing WT- or C481S-BTK treated with the indicated compounds for 16 hours. (B) TMD8 cells overexpressing either WT- or C481S-BTK were treated for 3 days with the indicated compounds. Antiproliferative effects of compounds were assessed using a CellTiter-Glo assay kit (Promega), and ED50 values were determined using GraphPad Prism nonlinear regression curve fit. (C) TMD8 cells were treated with the indicated compound combinations for 3 days. Plots show combination index (CI) scores. CI < 1 indicates synergy. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
BTK degraders overcome ibrutinib resistance in ABC DLBCL. (A) Immunoblots of TMD8 cells overexpressing WT- or C481S-BTK treated with the indicated compounds for 16 hours. (B) TMD8 cells overexpressing either WT- or C481S-BTK were treated for 3 days with the indicated compounds. Antiproliferative effects of compounds were assessed using a CellTiter-Glo assay kit (Promega), and ED50 values were determined using GraphPad Prism nonlinear regression curve fit. (C) TMD8 cells were treated with the indicated compound combinations for 3 days. Plots show combination index (CI) scores. CI < 1 indicates synergy. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Consistent with the activity of ibrutinib on other TEC family kinases, its activity against HCK (in addition to BTK) is responsible for antiproliferative effects in L265P-MYD88-driven ABC DLBCL cells.37 The parent inhibitor CGI1746 used for our degraders does not inhibit HCK,29 and our BTK degraders neither bind to nor degrade HCK (Figures 1C, 3A, and 4B). Thus, we hypothesized that we could recapitulate the therapeutic benefits of ibrutinib’s polypharmacology by combining BTK degradation with selective HCK inhibition. Consistent with this hypothesis, the HCK inhibitor A41925937 exhibited strong synergy with both DD-03-007 and DD-03-171 in TMD8 cells at numerous doses (Figure 3C). Moreover, DD-03-171 maintained synergy with A419259 in TMD8 cells that express C481S-BTK (supplemental Figure 1A). Thus, combining BTK degradation with HCK inhibition (or other selective kinase inhibitors) may be a viable therapeutic strategy with reduced off-target effects compared with ibrutinib alone.
![Figure 4. BTK degrader DD-03-171 shows on-target efficacy against MCL cells in vitro and in vivo. (A) Immunoblots from Mino cells treated as indicated for 18 hours; all bands for IKZF1 are specific. (B) Quantitative proteomics showing relative abundance of proteins in Mino cells treated with 200 nM DD-03-171 or DD-04-118 for 4 hours. (C) Mino cells were treated with the indicated compounds and concentrations for 3 days. Antiproliferative effects of compounds were assessed using the CellTiter-Glo assay (Promega), and ED50 values were determined using a GraphPad Prism nonlinear regression curve fit. (D) Immunoblot from mouse-depleted spleen samples of NSG mice engrafted with DFBL-96069 and treated for 3 days with the indicated compounds: vehicle (intraperitoneal [IP], twice daily), DD-03-171 (IP, daily, 50 mg/kg), lenalidomide (IP, daily, 50 mg/kg), ibrutinib (oral, daily, 30 mg/kg). All bands for IKZF1 are specific; each lane represents an individual tumor from different mice (3 mice per treatment cohort). (E) Tumor burden in mice treated for 2 weeks. Four to six hours after the last dose, peripheral blood was quantified for disease burden via flow cytometry. *P < .05. (F) Kaplan-Meier survival curve of mice treated as in panels D and E.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/133/9/10.1182_blood-2018-07-862953/4/m_blood862953f4.png?Expires=1765898324&Signature=SiH9IyNC2AA8AtJAZ15LUqtLKt2~8X6n9k1eaRzHFq7Z0HS2bkDEy~FE896cMnGy33pI-gcVMnGHXFek63~uNkzyAH6w6AG44aGuQ2wCPNV~ag8z81Ppg5RKxVoh7JgFdehc1-8j4G9~tDcnexNXJ4vvgGPnzdnl~yC-NeJkVI4Dmg5KwJEiPfCWJ~9vR~cvjK5y8v3Z266r1M8-tLE6Yx2iwfe5yKPn4eurM8VGB7jnw2WDe8ZlAObgEGgdnxw5akxRZi7-Yqsh6DPWimY96Z2hkbsdDkC7b0hJzWXldQyOtT2xXf7GW5PHFiRob8Nh0pLLWiPlQ2fSKk3yEeDu~w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
BTK degrader DD-03-171 shows on-target efficacy against MCL cells in vitro and in vivo. (A) Immunoblots from Mino cells treated as indicated for 18 hours; all bands for IKZF1 are specific. (B) Quantitative proteomics showing relative abundance of proteins in Mino cells treated with 200 nM DD-03-171 or DD-04-118 for 4 hours. (C) Mino cells were treated with the indicated compounds and concentrations for 3 days. Antiproliferative effects of compounds were assessed using the CellTiter-Glo assay (Promega), and ED50 values were determined using a GraphPad Prism nonlinear regression curve fit. (D) Immunoblot from mouse-depleted spleen samples of NSG mice engrafted with DFBL-96069 and treated for 3 days with the indicated compounds: vehicle (intraperitoneal [IP], twice daily), DD-03-171 (IP, daily, 50 mg/kg), lenalidomide (IP, daily, 50 mg/kg), ibrutinib (oral, daily, 30 mg/kg). All bands for IKZF1 are specific; each lane represents an individual tumor from different mice (3 mice per treatment cohort). (E) Tumor burden in mice treated for 2 weeks. Four to six hours after the last dose, peripheral blood was quantified for disease burden via flow cytometry. *P < .05. (F) Kaplan-Meier survival curve of mice treated as in panels D and E.
BTK degrader DD-03-171 shows on-target efficacy against MCL cells in vitro and in vivo. (A) Immunoblots from Mino cells treated as indicated for 18 hours; all bands for IKZF1 are specific. (B) Quantitative proteomics showing relative abundance of proteins in Mino cells treated with 200 nM DD-03-171 or DD-04-118 for 4 hours. (C) Mino cells were treated with the indicated compounds and concentrations for 3 days. Antiproliferative effects of compounds were assessed using the CellTiter-Glo assay (Promega), and ED50 values were determined using a GraphPad Prism nonlinear regression curve fit. (D) Immunoblot from mouse-depleted spleen samples of NSG mice engrafted with DFBL-96069 and treated for 3 days with the indicated compounds: vehicle (intraperitoneal [IP], twice daily), DD-03-171 (IP, daily, 50 mg/kg), lenalidomide (IP, daily, 50 mg/kg), ibrutinib (oral, daily, 30 mg/kg). All bands for IKZF1 are specific; each lane represents an individual tumor from different mice (3 mice per treatment cohort). (E) Tumor burden in mice treated for 2 weeks. Four to six hours after the last dose, peripheral blood was quantified for disease burden via flow cytometry. *P < .05. (F) Kaplan-Meier survival curve of mice treated as in panels D and E.
BTK degraders potently inhibit the proliferation of MCL cells
We next investigated the pharmacological effects of BTK degradation in MCL, a B-cell malignancy whose hallmark is the t(11:14) (q13:q32) chromosomal translocation between the cyclin D1 gene (CCND1) and the immunoglobulin heavy-chain locus (IGH@). Importantly, BTK is constitutively active in this disease, as a recent study found that despite similar BTK expression levels, levels of p-BTK (Y223), a surrogate marker of BTK activity, were higher in primary MCL cells than in resting B cells.38 As such, ibrutinib and the more selective BTK inhibitor acalabrutinib39 have shown beneficial effects in patients with MCL and have received US Food and Drug Administration approval for this indication. Interestingly, an ongoing clinical trial of ibrutinib, lenalidomide, and the anti-CD20 antibody rituximab has reported promising initial responses.40 As our degrader molecules contain moieties that have the potential to target both BTK and IKZF1/3, this presented us with the opportunity to determine the effects of codegradation of BTK and IKZF1/3 in MCL cells.
First, we investigated whether DD-03-171 retained the activity of its imide moiety on IKZF1/3 and found that it potently degraded IKZF1/3 as well as BTK in Mino MCL cells (Figure 4A). In contrast, DD-04-118, which only differs from DD-03-171 by a single atom (Figure 1A), had minimal effects on IKZF1/3 levels (Figure 4A).
To more broadly assess the effects of degrader treatment on cellular protein levels, we performed multiplexed mass spectrometry–based proteomic analysis of Mino cells following 4-hour treatment (Figure 4B). Using abundance measurements from TMT isobaric labels,41 whole-proteome analysis resulted in the identification and quantification of >125 000 unique peptides corresponding to >9000 proteins. Within the proteome, treatment with either DD-03-171 or DD-04-118 resulted in significant alterations in protein abundance of only 2 proteins each, and BTK was one of those proteins in both cases. Moreover, in DD-03-171–treated cells, IKZF1 and IKZF3 were degraded (∼1.5-fold change for both proteins, which did not meet our minimum twofold change threshold to be pictured on the scatterplot); in contrast, in DD-04-118–treated cells, IKZF1 and IKZF3 levels did not change (supplemental Table 4).
Next, we found that DD-03-171 had potent antiproliferative effects on Mino cells (ED50 = 12 nM; Figure 4C). Consistent with our finding that ibrutinib synergizes with lenalidomide to inhibit the proliferation of Mino cells (supplemental Figure 2), DD-03-171 was more potent than the “single-degrader” compound DD-04-118 (ED50 = 69 nM), the parent compound CGI1746, and the MCL-indicated, clinically approved drugs lenalidomide and ibrutinib. DD-03-171 also outperformed CGI1746 and ibrutinib in inhibiting the proliferation of Maver-1 MCL cells, but none of the 4 compounds had significant effects in Granta-519 MCL cells (supplemental Figure 3).
As the NF-κB pathway is critical in MCL cells,7 we examined the effects of BTK degradation on NF-κB signaling in Mino cells. As expected, targeted degradation of BTK resulted in reduced levels of active (phosphorylated) p65 NF-κB (supplemental Figure 4).
BTK degraders exhibit in vivo efficacy in MCL
Finally, we sought to investigate the effects of BTK degradation in vivo. To do so, we first compared the in vivo efficacy of DD-03-007 with DD-03-171 in terms of BTK degradation. Interestingly, while both compounds had similar pharmacokinetic profiles (supplemental Tables 2 and 3), only DD-03-171 induced significant degradation of BTK in nonmalignant mouse splenocytes (supplemental Figure 5).
After validating that DD-03-171 induced in vivo BTK degradation, we transitioned to in vivo PDX models of DLBCL (DFBL-18689) and MCL (DFBL-39435, DFBL-44685, DFBL-98848) (supplemental Table 5) engrafted into NSG mice. Treatment with DD-03-171 resulted in in vivo degradation of BTK in all models, while degradation of IKZF1 varied (supplemental Figures 6, 7A, 8A, and 9A). In DFBL-44685–engrafted mice, DD-03-171 treatment did not extend survival, which was not surprising, as neither ibrutinib nor lenalidomide treatment affected survival (supplemental Figure 8B). In contrast, in DFBL-39435–engrafted mice, DD-03-171 treatment and the combination of ibrutinib and lenalidomide similarly reduced circulating tumor burden after 14-day treatment as compared with the control cohort (supplemental Figure 7B). Moreover, in DFBL-98848–engrafted mice, the reduction in circulating disease in DD-03-171–treated mice was at least comparable to that seen in mice treated with ibrutinib, lenalidomide, or ibrutinib and lenalidomide in combination (supplemental Figure 9B).
Encouraged by these results, we compared the efficacy of DD-03-171 to ibrutinib and lenalidomide in DFBL-96069–engrafted mice. This model was derived from a patient who previously failed multiple rounds of therapy, including chemotherapy and ibrutinib. Immunoblots from tumor samples of a cohort of mice treated for 3 days revealed extensive degradation of BTK in purified DFBL-96069 cells (Figure 4D). While DD-03-171 efficiently degraded IKZF1/3 in vitro (Figure 4A), the in vivo situation was more complex, as BTK inhibition alone induced upregulation of IKZF1/3. This upregulation was reduced in the DD-03-171–treated cohort, suggesting that IKZF1/3 degradation did occur but was not sufficient to overcome the compensatory upregulation of these 2 proteins in response to loss of BTK signaling (Figure 4D). Finally, we observed significantly reduced tumor burden in the peripheral blood and a significant extension of survival in mice treated with DD-03-171 compared with the other 3 cohorts (Figure 4E-F).
Discussion
Previously, by converting a multitargeted kinase inhibitor into a degrader, we profiled the degradable kinome and identified BTK as a tractable target.14 Subsequently, several studies reported successful conversion of either a reversible ibrutinib scaffold42,43 or a different noncovalent analog derived from a previously published covalent BTK inhibitor44,45 into BTK degraders. Consistently, we found that CGI1746-based, CRBN-recruiting degraders with a variety of linker compositions potently induced cellular BTK degradation. Thus, BTK appears to be an easily degradable kinase.
Through rational linker engineering, we generated our lead compound, DD-03-171, which combines degradation of BTK, IKZF1, and IKZF3, 3 validated targets in B-cell malignancies. The combination of ibrutinib, lenalidomide, and rituximab has reported promising initial results,40 raising the question of whether concurrent degradation of BTK and IKFZ1/3 is advantageous over treating patients with the combination of an IMiD and a BTK inhibitor. Theoretically, targeting multiple proteins with a single molecule avoids potential adverse drug–drug interactions and offers more predictable pharmacokinetics than a drug combination, as well as simpler dosing for patients. We envision that this intentional polypharmacological approach of degrading IKZF1/3 along with a distinct oncogenic driver may have broad utility as a strategy for targeting multiple cancer vulnerabilities, particularly in B-cell malignancies.46
While several different BTK-binding ligands have been successfully converted into BTK degraders, it was unclear whether these compounds could efficiently induce BTK degradation in vivo and whether targeted BTK degradation would be a viable therapeutic modality. Here, we demonstrate that DD-03-171 has favorable pharmacokinetic and pharmacodynamic properties. To the best of our knowledge, this lead compound is the first targeted BTK degrader with demonstrated in vivo efficacy against B-cell malignancies. Nevertheless, there are several important factors to consider in weighing the relative risks and benefits of targeted BTK degradation. First, proteolysis-targeting chimeras may have suboptimal physicochemical properties for in vivo use. In a recent report of a BTK degrader, target degradation varied significantly between the spleen and lung despite similar distribution, potentially due to differences in the tissue-specific levels of CRBN.44 Second, ibrutinib has multiple off-target effects (eg, against HCK) that may contribute to its efficacy in specific contexts. Similarly, IMiDs are believed to have important effects independent of IKFZ1/3 degradation on the lymphoma microenvironment that contribute to their efficacy. It will be important to consider these off-target effects when designing studies using a triple degrader like DD-03-171 in select populations. Third, there are multiple mechanisms through which a lymphoma cell may be resistant to BTK degraders, including reduced uptake, increased efflux, mutations of BTK that block binding, activating mutations in PLCγ2 or alternative NF-κB,7 or alterations that affect E3 ligase/proteasome function. Finally, BTK is essential for normal B-cell development, and mice lacking IKZF1 have a complete block in B-cell differentiation at the pre-pro–B-cell stage.47 This suggests that a triple degrader could lead to profound B-cell lymphopenia akin to patients treated with anti-CD19 chimeric antigen receptor T cells. These patients may require long-term supplementation with IV immune globulin and have increased risk for reactivation of hepatitis B virus and other infections.
In conclusion, we developed highly potent and selective small-molecule degraders of BTK that exhibit a number of promising therapeutic characteristics, including (1) durable degradation and potent inhibition of signaling and antiproliferative effects, (2) an ability to overcome clinically relevant forms of ibrutinib resistance, and (3) efficacy in PDX models in vivo. Thus, targeted BTK degradation is a novel and promising therapeutic approach for treating B-cell malignancies, potentially even those resistant to conventional therapies.
The mass spectrometry raw data reported in this article have been deposited in the PRIDE Archive database (accession number PXD010568).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors gratefully acknowledge the patients who provided samples for these studies.
This work was supported by the Natural Sciences and Engineering Research Council of Canada (D.D.), Harvard University, the Dana-Farber Cancer Institute, the National Cancer Institute, National Institutes of Health (grant R01CA214608) (E.S.F), and a Damon Runyon Cancer Research Fellowship (DRG-2270-16) (E.S.W.). E.S.F. is a Damon Runyon-Rachleff Innovator supported in part by the Damon Runyon Cancer Research Foundation (DRR-50-18).
Authorship
Contribution: D.M.W., N.S.G., E.S.F., and S.P.T. conceived and designed the experiments; D.D. designed and synthesized all compounds; E.S.W. performed all immunoblotting and proliferation experiments, except those in TMD8 cells; S.M. and C.L. conducted the animal studies; R.P.N. performed docking analyses; K.A.D. performed the proteomics experiments; T.F. performed the biochemical assays; G.Y. prepared C481S-BTK–expressing cell models and performed immunoblots and antiproliferation experiments with TMD8 and HBL1 cells; Z.L. performed scale-up synthesis of DD-03-171 for animal studies; and D.D. and E.S.W. wrote the manuscript.
Conflict-of-interest disclosure: N.S.G. is a founder and equity holder in Syros, Soltego, Petra, and C4 Therapeutics. D.M.W. is a founder and equity holder in Travera and receives research support from AstraZeneca, Novartis, Aileron, Abbvie, Daiichi Sankyo, and Surface Oncology. E.S.F. is an equity holder and SAB member for C4 Therapeutics and receives research support from Novartis, and Astellas. The remaining authors declare no competing financial interests.
Correspondence: Nathanael S. Gray, Department of Cancer Biology, Dana-Farber Cancer Institute, Longwood Center, Room 2209, 360 Longwood Ave, Boston, MA 02215; e-mail: nathanael_fray@dfci.harvard.edu; and David M. Weinstock, Department of Medical Oncology, Dana-Farber Cancer Institute, Dana 510B, 450 Brookline Ave, Boston, MA 02215; e-mail: davidm_weinstock@dfci.harvard.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal