

Key Points
VWF p.Phe2561Tyr is the first gain-of-function variant, which increases the force sensitivity of VWF interaction with platelets.
VWF p.Phe2561Tyr is associated with repeated myocardial infarction, particularly in younger women.

Abstract
The frequent von Willebrand factor (VWF) variant p.Phe2561Tyr is located within the C4 domain, which also harbors the platelet GPIIb/IIIa-binding RGD sequence. To investigate its potential effect on hemostasis, we genotyped 865 patients with coronary artery disease (CAD), 915 with myocardial infarction (MI), and 417 control patients (Ludwigshafen Risk and Cardiovascular Health Study) and performed functional studies of this variant. A univariate analysis of male and female carriers of the Tyr2561 allele aged 55 years or younger revealed an elevated risk for repeated MI (odds ratio, 2.53; 95% confidence interval [CI], 1.07-5.98). The odds ratio was even higher in females aged 55 years or younger, at a value of 5.93 (95% CI, 1.12-31.24). Cone and plate aggregometry showed that compared with Phe2561, Tyr2561 was associated with increased platelet aggregate size both in probands’ blood and with the recombinant variants. Microfluidic assays revealed that the critical shear rate for inducing aggregate formation was decreased to 50% by Tyr2561 compared with Phe2561. Differences in C-domain circular dichroism spectra resulting from Tyr2561 suggest an increased shear sensitivity of VWF as a result of altered association of the C domains that disrupts the normal dimer interface. In summary, our data emphasize the functional effect of the VWF C4 domain for VWF-mediated platelet aggregation in a shear-dependent manner and provide the first evidence that a functional variant of VWF plays a role in arterial thromboembolism.
Introduction
Platelet adhesion and aggregation at the site of vessel injury is essential for primary hemostasis and is decisively mediated by von Willebrand factor (VWF) through its interaction with platelet membrane glycoprotein receptor GPIbα and integrin αIIbβ3 (GPIIb/IIIa) in the high shear arterial system and capillaries. VWF thus crucially contributes to the control of blood loss after vessel injury, but may, in contrast, induce a fatal step in the pathogenesis of myocardial infarction (MI) or stroke by thrombus formation.1,2 The role of the interaction between platelet glycoprotein receptors and VWF for primary hemostasis and for the pathogenesis of coronary artery disease (CAD) and MI has been addressed in a number of previous studies focusing on variant platelet glycoprotein alleles. The variant most often investigated, PIA1/A2, is located in GPIIIa of the GPIIb/IIIa receptor complex. Starting with Weiss` study in 1996,3 a number of studies followed, but with conflicting results.4-6 The majority of these studies assume an association of CAD and MI with the rare PIA2 allele.7 A possible explanation for the discrepancies observed was suggested by a study showing that the PIA1/A2 variant was related to premature MI, but not to CAD, indicating a role of distinct integrin genotypes in increased platelet thrombogenicity, but not in atherosclerosis.8 The significance of these results was underlined by functional studies showing increased platelet aggregability associated with the PIA2 allele.9
Epidemiological studies have shown that high levels of VWF are associated with an increased risk for arterial thromboembolism,10,11 whereas in patients with von Willebrand disease (VWD) a low incidence rate of arterial thrombosis has been observed.12 In addition, a deficiency of VWF was found to be protective against cerebral stroke in a VWF−/− murine model.13 Furthermore, reduced levels of ADAMTS13, the VWF-specific protease that proteolytically regulates the most functionally active high-molecular-weight VWF multimers, have been identified as significant risk factor for stroke and CAD.14,15
Apart from the merely elevated concentration of VWF observed in patients with arterial events, only genetic VWF variants affecting the level of circulating VWF have been linked to vascular events.16-18 However, no VWF gain-of-function (GOF) variants potentially implicated in thromboembolism have been described to date.
In a previous pilot study,19 we investigated the genetic variant c.7682T>A (p.Phe2561Tyr; rs35335161) in exon 45 of the VWF gene,20,21 for a potential association with MI in a group of 99 male patients with MI and 35 control patients. This variant is located in the VWF-C4 domain, which also harbors the VWF-RGD sequence (amino acids 2507-2509), the binding motif for platelet GPIIb/IIIa.22-24 Therefore, an influence of the Tyr2561 allele on the interaction between platelet GPIIb/IIIa and VWF in platelet aggregation appeared plausible. Although not statistically significant, we observed a higher frequency of Tyr2561 in the MI patient group. We here present evidence, both from a larger epidemiological study and particularly from functional studies, that the Tyr2561 allele is indeed a GOF variant, possibly associated with a higher risk for repeated MI in younger, and particularly in female, patients.
Methods
Study population and design
All participants in the study were characterized in detail for the established risk factors including hypercholesterolemia, smoking, diabetes, elevated fibrinogen, hypertension, weight, age, and sex. Precise definitions of these factors, inclusion and exclusion criteria, and a detailed description of the diagnosis of CAD and MI, as well as further information on the study design, can be found in Winkelmann et al.25
VWF genotypes (c.7682T/A), corresponding to Phe/Tyr2561-VWF, were assessed in 2197 white individuals undergoing coronary angiography for diagnostic purposes within the Ludwigshafen Risk and Cardiovascular Health Study (LURIC study; http://www.luric.online/). The entire study cohort consisted of 1780 patients with CAD (patients with at least 20% stenosis of at least 1 coronary arterial segment, irrespective of their individual MI status; includes 2 patients with previous MI but without CAD [1 man ≤55 years, 1 woman >55 years] and 417 control patients [patients with normal angiogram and without history of MI]). The study cohort was further subdivided as outlined in Table 1. Subgroups (≤55 years/>55 years) were defined in accordance with our previous study on the thrombogenicity of platelet GPIIIa variants PIA1/A2, which demonstrated an increased odds ratio (OR) in younger patients.8 Patients who were included in the study at the time they presented with their first or second MI and who were at that time aged 55 years or younger or older than 55 years were classified as 1 MI or more than 1 MI, age 55 years or younger or older than 55 years, respectively.
Number of controls and patients in the different subgroups
| Sex/Age (y) | Σ | Σ C | Σ C | Σ P | Σ P | CAD | Σ MI | 1MI | >1MI | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| All | All | Phe | Tyr | All | Phe | Tyr | Phe | Tyr | Phe | Tyr | Phe | Tyr | Phe | Tyr | |
| All | 2197 | 417 | 378 | 39 | 1780 | 1602 | 178 | 773 | 92* | 829 | 86 | 700 | 68 | 129 | 18 |
| All > 55 | 1551 | 277 | 248 | 29 | 1274 | 1152 | 122 | 660 | 77* | 492 | 45 | 435 | 41 | 57 | 4 |
| All < 55 | 646 | 140 | 130 | 10 | 506 | 450 | 56 | 113 | 15 | 337 | 41 | 265 | 27 | 72 | 14 |
| M, all | 1531 | 212 | 192 | 20 | 1319 | 1187 | 132 | 533 | 65* | 654 | 67 | 544 | 54 | 110 | 13 |
| M > 55 | 1012 | 115 | 102 | 13 | 897 | 810 | 87 | 434 | 53* | 376 | 34 | 329 | 31 | 47 | 3 |
| M < 55 | 519 | 97 | 90 | 7 | 422 | 377 | 45 | 99 | 12 | 278 | 33 | 215 | 23 | 63 | 10 |
| F, all | 666 | 205 | 186 | 19 | 461 | 415 | 46 | 240 | 27 | 175 | 19 | 156 | 14 | 19 | 5 |
| F > 55 | 539 | 162 | 146 | 16 | 377 | 342 | 35 | 226 | 24 | 116 | 11 | 106 | 10 | 10 | 1 |
| F < 55 | 127 | 43 | 40 | 3 | 84 | 73 | 11 | 14 | 3 | 59 | 8 | 50 | 4 | 9 | 4 |
| Sex/Age (y) | Σ | Σ C | Σ C | Σ P | Σ P | CAD | Σ MI | 1MI | >1MI | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| All | All | Phe | Tyr | All | Phe | Tyr | Phe | Tyr | Phe | Tyr | Phe | Tyr | Phe | Tyr | |
| All | 2197 | 417 | 378 | 39 | 1780 | 1602 | 178 | 773 | 92* | 829 | 86 | 700 | 68 | 129 | 18 |
| All > 55 | 1551 | 277 | 248 | 29 | 1274 | 1152 | 122 | 660 | 77* | 492 | 45 | 435 | 41 | 57 | 4 |
| All < 55 | 646 | 140 | 130 | 10 | 506 | 450 | 56 | 113 | 15 | 337 | 41 | 265 | 27 | 72 | 14 |
| M, all | 1531 | 212 | 192 | 20 | 1319 | 1187 | 132 | 533 | 65* | 654 | 67 | 544 | 54 | 110 | 13 |
| M > 55 | 1012 | 115 | 102 | 13 | 897 | 810 | 87 | 434 | 53* | 376 | 34 | 329 | 31 | 47 | 3 |
| M < 55 | 519 | 97 | 90 | 7 | 422 | 377 | 45 | 99 | 12 | 278 | 33 | 215 | 23 | 63 | 10 |
| F, all | 666 | 205 | 186 | 19 | 461 | 415 | 46 | 240 | 27 | 175 | 19 | 156 | 14 | 19 | 5 |
| F > 55 | 539 | 162 | 146 | 16 | 377 | 342 | 35 | 226 | 24 | 116 | 11 | 106 | 10 | 10 | 1 |
| F < 55 | 127 | 43 | 40 | 3 | 84 | 73 | 11 | 14 | 3 | 59 | 8 | 50 | 4 | 9 | 4 |
Given are the numbers and the distribution of Phe-only and Tyr-carriers, respectively, assuming a dominant model.
Σ MI, all patients with MI; 1MI, patients with exactly 1 MI; >1MI, patients with more than 1 MI; C, controls; CAD, patients with CAD/without MI; F, female; M, male; P, patients.
The subgroup includes 2 Tyr homozygous patients
Identification of the c.7682T>A (Phe2561Tyr) variant
DNA samples, isolated from peripheral blood cells using the commercial QIAmp DNA Blood Mini Kit (QIAGEN, Hilden, Germany) were from the LURIC study repository. Screening for the c.7682T>A variant was carried out by using the WAVE DNA Fragment Analysis System (Transgenomic Inc., New Haven, CT). Specificity of the resulting elution profile was confirmed by Sanger sequencing.
Statistical analysis
SAS software (version 8.1 by SAS Institute) was used for statistical analysis. Discrete variables were analyzed by means of the χ-square test, and in the case of smaller groups, also by means of the Fisher exact test. The Mann-Whitney U test (Wilcoxon rank sum test) was used to analyze continuous variables. Statistical significance was assumed at a P-value of ≤ .05.
The association of the Tyr2561 allele with CAD or MI was determined by stepwise multiple logistic regression analysis, controlling for established risk factors (age, sex, hypercholesterolemia [240 mg/dL or lipid-lowering medication], smoking [current or previous], hypertension [>140 mm Hg systolic or >90 mm Hg diastolic], diabetes mellitus, fibrinogen >360 mg/dL, overweight [BMI ≥ 26 kg/m2]). VWF:Ag was elevated above the normal range (50-160 U/dL), but was almost identical in patients (173 ± 72 U/dL) and controls (172 ± 69 U/dL). Heterozygous and homozygous carriers of the Tyr2561 allele were classified as Tyr2561-positive (dominant model), and carriers of only the Phe2561 allele as Tyr2561-negative. Only 2 patients (CAD without MI) were homozygous for Tyr2561. To test for an influence of the Tyr2561 allele on the average MI manifestation age, a multivariate regression analysis based on the Cox proportional hazard model was used. The event time was defined as the age at first and second MI (Phreg Procedure, SAS statistical package).
VWF parameters of probands
VWF:Ag and VWF collagen-binding activity (VWF:CB) was assessed in 18 healthy volunteers with Phe/Phe2561-VWF (VWF:Ag mean, 72 ± 15 U/dL; and VWF:CB mean, 85 ± 19 U/dL, respectively) and 13 Phe/Tyr2561 carriers (VWF:Ag, mean 113 ± 32 U/dL; and VWF:CB, mean 113 ± 36 U/dL, respectively).
Cone and plate aggregometry
Cone and plate analysis (CPA) was performed with citrated whole blood from the probands described here, employing the IMPACT-R device (Matis Medical Inc., Beersel, Belgium). Platelet adhesion (surface coverage) and aggregate size were analyzed by the internal IMPACT-R software.26,27 For details, see supplemental Methods, available on the Blood Web site.
For studies with recombinant (r)VWF, plates were precoated with 50 µg/mL of rPhe2561-VWF, incubated for 3 hours at room temperature, and washed twice with PBS. Subsequently, 130 µL of Phe2561-VWF whole blood supplemented with either rPhe2561-VWF or rTyr2561-VWF to a final concentration of 50 µg/mL were used for CPA to determine platelet aggregate size and surface coverage. CPA was performed at shear rates of 1000, 1800, and 3000 s−1 for 2 minutes.
Microfluidic assays
Functional characterization of VWF-induced collective network formation28,29 under high-shear conditions was performed in air-pressure-driven microfluidic channels (Bioflux, San Francisco, CA; width, 350 µm; height, 70 µm) coated with 50 µg/mL of either rPhe2561-VWF or rTyr2561-VWF, as previously described.28,30 Citrated whole blood was washed and platelets fluorescently stained as published. Washed blood, including 200 000/µL platelets, 45% washed hematocrit, and 10 µg/mL of either rPhe2561-VWF or rTyr2561-VWF, was subjected to various shear rates in the range of 500 to 5000 s−1 (with a nominal shear rate precision of 36 s−1) . Live cell fluorescence movies were taken with 8 frames per second. At least 3 independent experiments were performed for each group. For image analysis, we used the ZEN software (Zeiss AG, Jena, Germany) and the open-source software ImageJ (V. 1.46r, National Institutes of Health, Bethesda, MD).31
Static VWF-platelet receptor binding assays
Comparative binding of rPhe2561-VWF and rTyr2561-VWF to GPIIb/IIIa was assessed by a static assay using GPIIb/IIIa isolated from platelets, as previously described.32
In addition, we performed a cell-based static assay, detecting the binding of HEK293 cells stably expressing a constitutively active mutant of the GPIIb/IIIa complex33 to immuno-adsorbed VWF in 96-well plates. For details, see supplemental Methods.
Ristocetin-induced GPIbα-binding to recombinant Phe2561, Tyr2561, and the VWF type 2B mutant p.Ile1309Val, respectively, was measured by an enzyme-linked immunosorbent assay, essentially as described (supplemental Methods).34
Expression of recombinant full-length VWF and VWF fragments
HEK293 cell lines, stably expressing full-length Phe2561-VWF, Tyr2561-VWF, or both alleles after cotransfection, were derived from transiently transfected HEK293 cell clones,35 selected by 500 µg/mL G418 in cell culture medium.
To express VWF domain constructs, the VWF signal peptide and residues 1 to 3 of the propeptide were cloned into the pIRES vector, using restriction sites NheI and EcoRI. PCR-amplified VWF domain sequences C3-C4 (residues 2426-2581), C3-CTCK (residues 2426-2813), and C1-CTCK (residues 2256-2813), respectively, carrying either Phe- or Tyr2561, all with a hexapolyhistidine-tag and stop codon sequence, were cloned C-terminally of the signal peptide sequence using restriction sites EcoRI and BamHI. Primers are available on request. Protein expression was performed as previously described.35 The fragments of rPhe2561-VWF and rTyr2561-VWF (ie, VWF domains C3-C4, C3-CTCK, and C1-CTCK, respectively) were purified using a Nickel sepharose column. The rPhe2561- and rTyr2561-VWF C4-domain fragments were expressed and purified as described in supplemental Methods. Protein concentrations were determined on a UV2101PC spectrophotometer (Shimadzu, Duisburg, Germany). The extinction coefficients for all constructs were calculated from the number of tyrosine and tryptophan residues of the respective sequences, using extinction coefficients of 1215 L⋅mol−1⋅cm−1 for tyrosine and 5630 L⋅mol−1⋅cm−1 for tryptophan residues.36
Near- and far-UV circular dichroism spectra
Mean residue molar ellipticity corrected for the signal of the buffer were measured for both Phe2561 and Tyr2561 in constructs C4, C3C4, C3CTCK, and C1CTCK at 20°C on an Aviv Biomedical Model 420SF circular dichroism spectrometer (Aviv Biomedical, Lakewood, NJ). Far-ultraviolet (UV) circular dichroism (CD) spectra (200-260 nm, 1-nm bandwidth) in a 1-mm quartz cell were averaged during a 60-second integration time. Near-UV CD spectra (260-360 nm, 1-nm bandwidth) in a 2-mm quartz cell (C4) and a 10-cm cylindrical quartz cell (C3C4, C3CTCK, and C1CTCK) were averaged during a 120-second integration time.
The study was conducted in conformity to the Declaration of Helsinki37 and to the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use Guidelines (https://www.ich.org/home.html, accessed October 2017). The LURIC study was approved by the ethics committee at the Ärztekammer Rheinland-Pfalz (Mainz, Germany). Informed consent was obtained from all individual participants included in the study. Testing healthy volunteers was approved by the Ethics Committee of the Medical Faculty Mannheim, Heidelberg University (Mannheim, Germany). Appropriate informed consent was obtained from all subjects.
Results
Prevalence and relation of established risk factors to the Tyr2561 allele in the study population
Hypercholesterolemia, smoking, arterial hypertension, hyperfibrinogenemia, diabetes mellitus, and overweight were statistically significant risk factors in the CAD/MI study population both in a univariate and in a multivariate logistic regression analysis in the cohort of patients compared with controls from the LURIC study.8 Although present at an elevated frequency in the entire patient group (CAD without MI, 1 MI, and >1 MI), the Tyr2561 allele did not appear as a statistically significant risk factor compared with the control group (univariate OR, 1.12 [95% confidence interval [CI], 0.77-1.61]; multivariate OR, 1.33 [95% CI, 0.88-2.0]).
In the multivariate analysis of the subgroup with repeated events (>1 MI), the presence of Tyr2561 reached statistical significance and a relative risk of 2.78 compared with the control patients (95% CI, 1.12–6.5).
Results of the univariate analysis with ORs and CIs of all subgroups show that in patients aged 55 years or younger, Tyr2561 carriers had an OR of 2.53 (95% CI, 1.07-5.99) to experience more than 1 MI compared with the control patients (Figure 1). In the sex-specific analysis of this group, the risk was confirmed for females (OR, 5.93; 95% CI, 1.12-31.24), whereas for males the increased risk was present, but not statistically significant (OR, 2.04; 95% CI, 0.74-5.65).
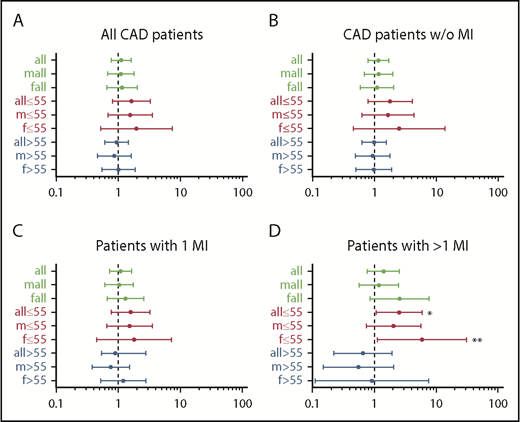
Forest plots of odds ratios for Tyr2561-VWF in the different patient groups relative to control patients. (A) All patients with CAD, (B) all patients with CAD without MI, (C) all patients with 1 MI, and (D) all patients with more than 1 M1. Asterisks point to significant results in patients aged 55 years or younger with more than 1 MI: *OR, 2.53 (95% CI, 1.07-5.98); **OR, 5.93 (95% CI, 1.12-31.24).
Forest plots of odds ratios for Tyr2561-VWF in the different patient groups relative to control patients. (A) All patients with CAD, (B) all patients with CAD without MI, (C) all patients with 1 MI, and (D) all patients with more than 1 M1. Asterisks point to significant results in patients aged 55 years or younger with more than 1 MI: *OR, 2.53 (95% CI, 1.07-5.98); **OR, 5.93 (95% CI, 1.12-31.24).
In contrast, comparing the group older than 55 years/>1 MI with comparative control patients, both in the entire study populations and in the female and male subgroups, Tyr2561 carriers were underrepresented in the patient group; however, the difference did not prove statistically significant. As a consequence, we analyzed the age-dependent influence of the Tyr2561 allele on the manifestation of more than 1 MI, using a multivariate Cox proportional hazard model. Whereas in the multivariate analysis the effect was not significant in males (P = .11), we observed a significant difference in event-free survival between female noncarriers and carriers of Tyr2561 who experienced a second MI significantly earlier than Phe2561 carriers (P = .024), and the time between the first and second MI was significantly shorter in carriers of Tyr2561 (P = .013; mean age at second MI, 64.9 vs 51.9 years in women and 59.6 vs 53.7 years in men). The univariate analysis gave significant differences in all subgroups (Figure 2).

Lifetable analysis of patients with more than 1 MI. (A-C) The proportion of patients with a second MI is presented on the ordinate, the age at onset of the second MI is presented on the abscissa. (D) Time between first and second MI in patients with Tyr2561 compared with Phe2561. Significant differences at P < .05.
Lifetable analysis of patients with more than 1 MI. (A-C) The proportion of patients with a second MI is presented on the ordinate, the age at onset of the second MI is presented on the abscissa. (D) Time between first and second MI in patients with Tyr2561 compared with Phe2561. Significant differences at P < .05.
Functional studies of Tyr2561-VWF
In a static assay, we first measured binding of VWF variants to GPIIb/IIIa isolated from platelets. Binding of rTyr2561-VWF, coexpressed with rPhe2561-VWF, was slightly enhanced when compared with rPhe2561-VWF alone (Figure 3A). This effect was absent when measuring only rTyr2561-VWF. Next, we used our cell-based GPIIb/IIIa binding assay, which determines binding to cell-membrane-incorporated, constitutively active GPIIb/IIIa. Here, no significant difference was observed between VWF variants (Figure 3B). In both assays, the p.Asp2509Glu control variant with inactivated RGD motif exhibited almost no binding, as expected.
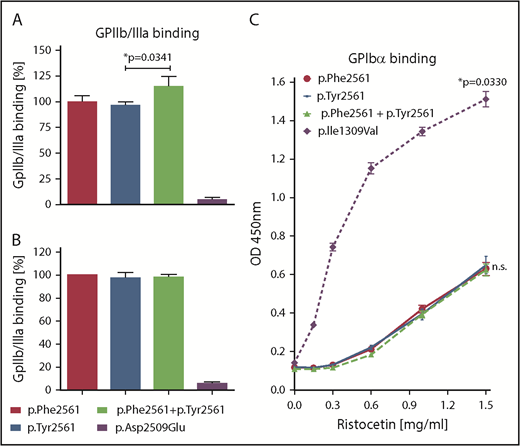
Assessment of VWF-GPIIb/IIIa and GPIbα binding under static conditions. (A) rVWF variants were incubated in microtiter wells with immunoadsorbed GPIIb/IIIa purified from platelets. Bound VWF was detected using an HRP-conjugated anti-VWF antibody. Presented is the ratio relative to Phe2561. (B) Immunoadsorbed rVWF variants were incubated with HEK293 cells stably expressing a constitutively active mutant of GPIIb/IIIa. Bound cells were detected using mouse anti-GPIIb/IIIa and an HRP-coupled secondary antibody polyclonal goat anti-mouse/HRP. (C) rVWF variants were incubated in microtiter wells with immunoadsorbed recombinant wild-type GPIbα fragment (amino acids 1-285) at indicated Ristocetin concentrations. Detection of bound VWF was performed employing a rabbit anti-human VWF antibody and an HRP-coupled secondary antibody polyclonal goat anti-rabbit/HRP. The data (A-C) represent the mean values ±SD of 3 independent measurements. Unpaired t-test, *P < .05; n.s., not significant.
Assessment of VWF-GPIIb/IIIa and GPIbα binding under static conditions. (A) rVWF variants were incubated in microtiter wells with immunoadsorbed GPIIb/IIIa purified from platelets. Bound VWF was detected using an HRP-conjugated anti-VWF antibody. Presented is the ratio relative to Phe2561. (B) Immunoadsorbed rVWF variants were incubated with HEK293 cells stably expressing a constitutively active mutant of GPIIb/IIIa. Bound cells were detected using mouse anti-GPIIb/IIIa and an HRP-coupled secondary antibody polyclonal goat anti-mouse/HRP. (C) rVWF variants were incubated in microtiter wells with immunoadsorbed recombinant wild-type GPIbα fragment (amino acids 1-285) at indicated Ristocetin concentrations. Detection of bound VWF was performed employing a rabbit anti-human VWF antibody and an HRP-coupled secondary antibody polyclonal goat anti-rabbit/HRP. The data (A-C) represent the mean values ±SD of 3 independent measurements. Unpaired t-test, *P < .05; n.s., not significant.
Direct influence of rTyr2561-VWF on platelet GPIbα-binding affinity under static conditions could be excluded, as there was no significant difference in Ristocetin-induced GPIbα-binding between Phe2561 and Tyr2561, in contrast to significantly enhanced GPIbα- binding of the common VWD type 2B mutant p.Ile1309Val (Figure 3C).
To determine a potential effect of the Tyr2561 allele on platelet aggregation under shear flow conditions, we measured platelet aggregation in whole blood of Tyr2561-VWF carriers compared with homozygotes of the Phe2561 allele. CPA analysis revealed an increase in aggregate size by about 50% for Tyr2561 (Figure 4A, C-E, blue bars) compared with the Phe2561 allele (Figure 4B, C-E red bars), which was accompanied by an increase in surface coverage (Figure 4F-H).
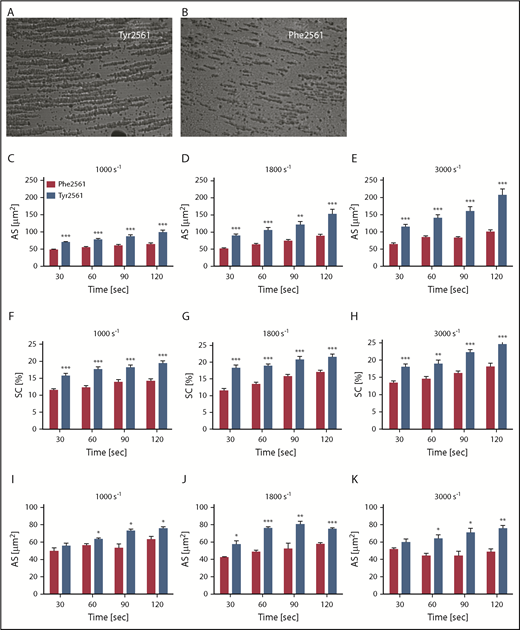
Assessment of VWF-GPIIb/IIIa binding under flow conditions. (A-B) CPA of platelet aggregation under flow conditions. (C-H) CPA of whole blood from volunteers carrying either the Phe2561 (n = 18) or the Tyr2561 allele (n = 13). (I-K) Phe2561 whole blood supplemented with 50 µg/mL of either recombinant Phe2561 (red bars) or Tyr2561 (blue bars). Aggregate size (C-E,I-K) and surface coverage (F-H) were determined at 1000 s−1, 1800 s−1, and 3000 s−1 at the indicated points, aggregates were stained using May-Grünwald solution. Seven pictures per test were recorded employing a built-in microscope and camera and analyzed using the IMPACT-R image analyzer software. Each test was performed in duplicate at all points. Mean values ± SEM are shown. Asterisks indicate a significant difference between controls and Tyr2561 at each point (unpaired t-test, *P < .05; **P < .005; ***P < .0001).
Assessment of VWF-GPIIb/IIIa binding under flow conditions. (A-B) CPA of platelet aggregation under flow conditions. (C-H) CPA of whole blood from volunteers carrying either the Phe2561 (n = 18) or the Tyr2561 allele (n = 13). (I-K) Phe2561 whole blood supplemented with 50 µg/mL of either recombinant Phe2561 (red bars) or Tyr2561 (blue bars). Aggregate size (C-E,I-K) and surface coverage (F-H) were determined at 1000 s−1, 1800 s−1, and 3000 s−1 at the indicated points, aggregates were stained using May-Grünwald solution. Seven pictures per test were recorded employing a built-in microscope and camera and analyzed using the IMPACT-R image analyzer software. Each test was performed in duplicate at all points. Mean values ± SEM are shown. Asterisks indicate a significant difference between controls and Tyr2561 at each point (unpaired t-test, *P < .05; **P < .005; ***P < .0001).
To confirm that the observed effect is specifically induced by VWF, we further performed the CPA experiments described here, using whole-blood samples supplemented with either 50 µg/mL rPhe2561-VWF or rTyr2561-VWF. The addition of rVWF-Tyr2561 significantly increased aggregate size at all shear rates applied compared with the addition of rPhe2561 (Figure 4I-K).
Assessment of shear-dependent VWF function by the Bioflux system
For a complementary description of the shear-dependent effect of Tyr2561, we performed microfluidic assays by the Bioflux system (Figure 5). At a low shear rate of 500 s−1, only single platelets were rolling along the VWF biofunctionalized surface. In the presence of rPhe2561-VWF, we first observed platelet-decorated VWF fibers at the channel surface at shear rates of 2000 s−1, increasing in size of about hundred micrometers. Above a critical shear rate of 4000 s−1, these reversibly formed fiber-like structures partially detach and induce the origin of large VWF-platelet collective network aggregates rolling along the surface.29,30 Whereas we did not detect differences in platelet adhesion at low shear rates (500 s−1) on rPhe2561-VWF and rTyr2561-VWF biofunctionalized surfaces (supplemental Figure 1), in the presence of rTyr2561-VWF, the critical shear rate for collective network formation was significantly decreased to 2000 s−1. These data confirm a GOF effect by Tyr2561-VWF (supplemental Movie 1). As expected, neither platelet binding fibers nor collective networks could be found in the absence of the plasmatic VWF fraction (data not shown).
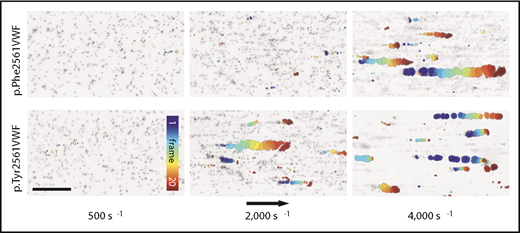
VWF-induced collective network formation of rPhe2561 and rTyr2561 VWF at indicated shear rates. Each image represents a composition of 20 sequential frames of a live-cell fluorescence movie taken at a frequency of 8 frames per second. By subtracting identical pixels among frames, a color-coded addition of these differential images from frame 0 (blue) to frame 20 (red) along the color scale, combined with the inverted gray-scaled background image of the start frame, allows precise detection and motion tracking of VWF-induced collective networks. Therefore, only moving networks are displayed in color enabling an exact determination of the critical shear rate. The rTyr2561-VWF variant (lower row) shows a significantly decreased critical shear rate in comparison with rPhe2561-VWF (upper row) to less than 50%. The black arrow indicates the flow direction and a scale bar corresponding to 100 µm.
VWF-induced collective network formation of rPhe2561 and rTyr2561 VWF at indicated shear rates. Each image represents a composition of 20 sequential frames of a live-cell fluorescence movie taken at a frequency of 8 frames per second. By subtracting identical pixels among frames, a color-coded addition of these differential images from frame 0 (blue) to frame 20 (red) along the color scale, combined with the inverted gray-scaled background image of the start frame, allows precise detection and motion tracking of VWF-induced collective networks. Therefore, only moving networks are displayed in color enabling an exact determination of the critical shear rate. The rTyr2561-VWF variant (lower row) shows a significantly decreased critical shear rate in comparison with rPhe2561-VWF (upper row) to less than 50%. The black arrow indicates the flow direction and a scale bar corresponding to 100 µm.
Conformation studies of Tyr2561-VWF
To identify conformational effects possibly caused by the Tyr2561-VWF variant at the level of the platelet integrin binding VWF C4 domain, its high-resolution structure was determined in a separate contribution (for details, see Xu et al38 ). The structure shows that the site of Phe2561 is remote from the integrin RGD binding site, and no significant change in either the overall conformation of the C4 domain or its ability of binding to platelet integrin could be detected. These data suggest that the VWF-GOF by Tyr2561 may, rather, be attributed to effects by structural perturbations from neighboring parts of the VWF assembly.
To address this question, we used CD spectroscopy to compare Phe2561 with the Tyr2561 variant in VWF constructs of various lengths and complexity, including the separate C4 domain, identical to the construct used for three-dimensional structure determination (for details, see supplemental Methods and Xu et al38 ), C3C4 covering a tandem of VWF domains, and extended VWF constructs that include the C-terminal CK domain (C3-CTCK, C1-CTCK). When the CK domain that mediates C-terminal VWF dimerization is present, VWF is dimeric, as expected, whereas C4 and C3C4 are monomeric. In agreement with the accompanying paper on the structure of the C4 domain,38 the spectral minimum at 208 nm in the far-UV spectra of C4, C3C4, C3-CTCK, and C1-CTCK indicates that these C domains are a mixture of β-sheet and random coil secondary structure (Figure 6A-D, left panels). Also supported by the C4-domain structure, the far-UV spectrum of Tyr2561 is virtually identical to that of Phe2561 for the monomeric C4 domain and the C3C4 domain pair. In contrast, Tyr2561 in the dimeric C3CTCK-domain and C1CTCK-domain constructs shows a detectable loss of CD around 230 nm relative to Phe2561. As this change is only detected in the presence of the C-terminal CK domain, Tyr2561 appears to have only an effect in the C-terminally assembled coiled-coil structure of the VWF C-domain segment in which local structural changes near Phe2561 could lead to disturbance of the assembly, and ultimately lower the overall content of ordered secondary structure. The significant near-UV CD spectral signals indicate that all C-domain constructs remain folded into a well-defined tertiary structure.
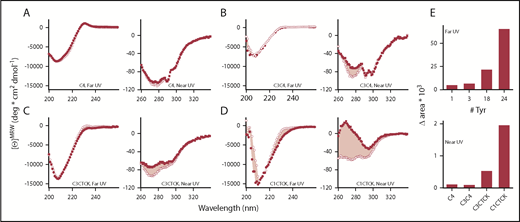
CD spectra for Phe2561- (•,▪) and Tyr2561-VWF (○,□). (A) C4, (B) C3C4, (C) C3CTCK, and (D) C1CTCK. Near-UV (squares) and far-UV CD spectra (circles) are shown on the left side and right side of each panel, respectively. The pink shaded area indicates the area difference between Phe2561-VWF and Tyr2561-VWF in each construct. This difference (Δarea) is plotted in panel E as a function of the number of tyrosine residues in each protein construct.
CD spectra for Phe2561- (•,▪) and Tyr2561-VWF (○,□). (A) C4, (B) C3C4, (C) C3CTCK, and (D) C1CTCK. Near-UV (squares) and far-UV CD spectra (circles) are shown on the left side and right side of each panel, respectively. The pink shaded area indicates the area difference between Phe2561-VWF and Tyr2561-VWF in each construct. This difference (Δarea) is plotted in panel E as a function of the number of tyrosine residues in each protein construct.
To further emphasize these spectral differences, Figure 6E illustrates that the area between Phe-VWF and Tyr2561-VWF spectra increases in both the far-UV and near-UV wavelength range as more of the C domains are added in the recombinant C-domain constructs. Plotted as a function of the number of tyrosines present in each of the constructs, the spectral differences of Tyr2561 occur primarily in the dimeric C-terminal C3-CTCK and C1-CTCK, suggesting that the observed conformational effect is not merely an intrinsic spectral property of tyrosine but, rather, representative of the combined effect of 2 tyrosines (1 in each C4 domain of the dimer) on the overall C-terminal domain organization of VWF. Taken together, these comparative spectroscopic results indicate that Tyr2561 causes a small perturbation of the secondary structure that propagates to an altered tertiary structure of the C domains, which might induce altered interactions between C domains in the dimeric constructs.
Discussion
Previous studies on the role of VWF in MI and stroke were mainly focusing on its plasma concentration.10,29 Only recently genetic variants were found that were associated with higher VWF:Ag levels.16 These variants seem to play a role in MI,18 but not in stroke.17 However, no systematic functional studies of common nonsynonymous VWF variants, possibly correlating with a prothrombotic state, have been reported before.
We have investigated a common variant of VWF, the Tyr2561 allele in the VWF C4 domain, which harbors the binding site for GPIIb/IIIa, in a cohort of patients with CAD and MI and in corresponding control patients. We showed that among patients with recurrent MI, the Tyr2561-VWF variant is more common compared with a control group, reaching statistical significance in younger individuals, particularly in younger women. However, because of the number of subgroups, the overall epidemiological results are only suggestive, and the results require confirmation by a larger prospective study. Nevertheless, we hypothesize that the variant is associated with increased thrombogenicity, as indicated by consistently elevated odds ratios higher than 1 in younger patients (≤55 years) compared with odds ratios lower than 1 in older individuals. As a consequence, patients with Tyr2561 having their MI at a younger age cannot become part of the older cohort later in their life, when conventional risk factors are playing a more important role. This is also in line with the lifetable analysis of Tyr2561 carriers, showing a significantly earlier onset of MI, particularly in recurrent MI (Figure 2).
The usual in vitro standard assays (PFA 100, VWF:Ag, VWF:CB, VWF multimer analysis) failed to detect increased quantitative or qualitative parameters of the variant, and as a result of analyzing only small subgroups, statistical evaluation was compromised. To overcome the statistical limitations of our clinical study and to support our GOF hypothesis, we functionally characterized Tyr2561-VWF compared with Phe2561-VWF in carriers of the respective variants by 3 static and 2 shear-based assays.
In 1 static assay, using isolated GPIIb/IIIa from platelets, we found no enhanced binding for rTyr2561-VWF, but a slightly enhanced binding for rTyr2561-VWF coexpressed with rPhe2561-VWF. This points to some enhanced binding of the heterozygous variant, even without the influence of shear. However, we could not confirm these results by using our cell-based binding assay with constitutively active GPIIb/IIIa incorporated in the cell membrane. We think that the cell-based assay is possibly more reliable, as it is closer to the physiological situation, compared with the assay with isolated GPIIb/IIIa, extracted from the platelet membrane. Furthermore, residue 2561 and the RGD sequence are in remote positions in the C4-domain structure.38 Therefore, we favor the interpretation that, under static conditions, differences in binding between rTyr2561-VWF and rPhe2561-VWF to GPIIb/IIIa will rather be negligible, speaking against a higher affinity of rTyr2561-VWF to GPIIb/IIIa. In addition, although not unexpected, we excluded a higher affinity of GPIbα to Tyr2561-VWF as an alternative mechanism.
In contrast, under shear conditions measured by CPA, platelet aggregate size in the blood of all Tyr2561 probands was significantly increased at all shear conditions compared with homozygous Phe2561 carriers. Although the mean of plasma VWF:Ag in the Tyr2561 carriers was higher than in the Phe2561 carriers, the rather homogenous results of CPA measurements with a very small standard deviation, despite the variable VWF:Ag in both groups, speaks against a dosage effect. Furthermore, using recombinant Tyr2561-VWF, we confirmed the specificity of the effect for VWF.
The recombinant variant also causes a marked decrease in the critical shear rate for collective network formation of VWF and platelets to less than 50%, which provides complementary evidence for a GOF of Tyr2561-VWF under high-shear conditions and is in accordance with our clinical data.
A possible explanation for the enhanced shear-sensitivity of VWF is provided by the observed conformational changes induced by Tyr2561: the comparative CD spectroscopy demonstrates that there is an intrinsic effect of Tyr2561 on the secondary and tertiary structure of the C domains in the presence of the C-terminal CK dimerization modules. The conformational effect of Tyr2561 to increase the propensity for local disorder of the peptide backbone is magnified in the dimeric C3CTCK and C1CTCK constructs, resulting in enhanced changes in tertiary structure that alter domain/domain interactions within each VWF monomer. In this case, the hydrodynamic force required for opening of the VWF stem would be decreased because of a lower free energy barrier required for the globular-to-extended transition under shear stress. These changes may lead to alterations in VWF-dimer assembly and destabilization of the “bouquet” arrangement39 of the entire VWF-stem region. In principle, this could facilitate opening VWF to a more extended conformation, resulting in a larger effective length of VWF multimers that increases the exposure of A1 domains and/or GPIIb/IIIa binding sites in the C4 domains and could also lead to enhanced VWF self-association which may amplify these effects.40,41
Similar as discussed for other prothrombotic variants of the hemostatic system such as factor V Leiden, Tyr2561-VWF might provide an evolutionary advantage by protection against excessive blood loss in risk situations, against the background of a relatively mild prothrombotic state, which would explain its relatively high frequency in the normal population.
Our findings emphasize the complex functional impact of the VWF-C domains in general, not only for patients with thromboembolism like MI or stroke but also for patients with unexplained mucosal bleeding disorders resembling VWD, which might remain undiagnosed by only applying the standard diagnostic tests. Shear-based binding assays for routine diagnostic purposes could fill this diagnostic gap.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank P. Legendre for performing the static VWF-GPIIb/IIIa binding assay, Gesa König for excellent technical support, and Achim Löf for very helpful discussions.
The work was financially supported by the German Research Foundation to the Research Group FOR1543 (SHENC; R.S., M.A.B., U.K., T.O., V.H., S.W.S., E.-R.X., M.W.), funding #Schn 325/6-2, and National Institutes of Health, National Heart, Lung, and Blood Institute grant HL109109 (M.A.).
Authorship
Contribution: R.S. designed and directed research, supervised experiments, analyzed experimental data, and wrote the manuscript; N.H. performed genetic testing and established the genetic database of the study population; M.A.B. designed and supervised functional studies; U.K. performed and analyzed CPA experiments; T.O. developed and expressed recombinant genetic constructs; V.H. and S.W.S. carried out and analyzed the microfluidic experiments; C.V.D. evaluated the VWF-GPIIb/IIIa assay; A.T. and M.A. carried out CD studies; W.M. established the LURIC study and provided patient data; E.-R.X. provided rVWF C4-domain constructs; M.W. helped in interpreting the biophysical data in the context of high-resolution structural data; R.B.Z. carried out the statistical evaluation and wrote the respective part of the study; and all authors contributed to the editing of the manuscript and approved the final version.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Reinhard Schneppenheim, University Medical Center Hamburg-Eppendorf, Department of Pediatric Hematology and Oncology, Martinistr 52, 24105 Hamburg, Germany; e-mail: schneppenheim@uke.de.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal