

Key Points
Platelet GPVI is a key regulator of vascular integrity in growing tumors.
Inhibition of platelet GPVI induces tumor hemorrhage and increases efficacy of chemotherapeutics in mice.

Abstract
Maintenance of tumor vasculature integrity is indispensable for tumor growth and thus affects tumor progression. Previous studies have identified platelets as major regulators of tumor vascular integrity, as their depletion selectively rendered tumor vessels highly permeable and caused massive intratumoral hemorrhage. While these results established platelets as potential targets for antitumor therapy, their depletion is not a treatment option due to their essential role in hemostasis. Thus, a detailed understanding of how platelets safeguard vascular integrity in tumors is urgently demanded. Here, we show for the first time that functional inhibition of glycoprotein VI (GPVI) on the platelet surface with an antibody (JAQ1) F(ab)2 fragment rapidly induces tumor hemorrhage and diminishes tumor growth similar to complete platelet depletion while not inducing systemic bleeding complications. The intratumor bleeding and tumor growth arrest could be reverted by depletion of Ly6G+ cells, confirming them to be responsible for the induction of bleeding and necrosis within the tumor. In addition, JAQ1 F(ab)2–mediated GPVI inhibition increased intratumoral accumulation of coadministered chemotherapeutic agents, such as Doxil and paclitaxel, thereby resulting in a profound antitumor effect. In summary, our findings identify platelet GPVI as a key regulator of vascular integrity specifically in growing tumors and could serve as a basis for the development of antitumor strategies based on the interference with platelet function.
Introduction
Systemic cancer therapy is often hampered by dose-limiting effects, which considerably decreases the efficacy of applied chemotherapeutic agents. One of the major drawbacks of conventional anticancer therapy is that IV administrated cytotoxic drugs usually do not selectively accumulate in tumor tissues.1,2 Cytotoxic drugs and targeted therapeutics accumulate at significantly lower doses in solid tumors than in nontarget organs, increasing side effects and the risk that treatment has to be terminated prematurely.3-6 Thus, development of selective anticancer strategies to increase accumulation of drugs within solid tumors represents an important goal in oncology research.
The highly dysfunctional vasculature caused by the chronically inflamed tumor microenvironment and its highly proangiogenic profile is a major reason for the reduced transport of therapeutics into the tumor. It has therefore long been speculated that antiangiogenic drugs could improve tumor drug supply.7 However, experimental evidence indicates that in many cases, antiangiogenic drugs actually further inhibit drug transport by reducing vascular density.8-11 This would be in line with the intended purpose of these agents to starve the tumor by depriving it of its ability to initiate the growth of new supplying blood vessels.12-14
Platelets are small anucleate cells critical for primary hemostasis but also maintenance of vascular integrity under certain pathological conditions such as inflammation. Depletion of platelets in tumor-bearing mice has been shown to induce the loss of vascular integrity and thus profound bleeding in the tumor while leaving vessels in nontumor tissue largely unaffected.15 Interestingly, it has been shown that platelet depletion promotes the intratumoral accumulation of chemotherapeutic agents, thereby enhancing the antitumoral effect without affecting its overall toxicity.15 These studies revealed a crucial tumor-supporting role of platelets and indicated that targeting platelets might be a valid strategy to limit tumor progression and potentiate the efficacy of chemotherapy. While effective in experimental animals, the induction of acute thrombocytopenia in cancer patients is not a therapeutic option due to severe side effects on hemostasis. Thus, it is imperative to identify the molecular mechanisms how platelets safeguard vascular integrity in tumors in order to develop antiplatelet agents allowing selective destabilization of the tumor vasculature during chemotherapy in patients without triggering unwanted systemic bleeding complications.
The platelet receptor for collagen, laminin and fibrin, glycoprotein VI (GPVI) is a 58- to 60-kDa type I transmembrane protein that centrally regulates multiple platelet functions, including adhesion, activation, aggregation, and procoagulant activity.16-21 For years, GPVI has been considered as a potentially safe antithrombotic target based on the observation that its loss or functional inhibition provides profound protection in models of arterial thrombosis while having only very limited effects on normal hemostasis.22 Besides its central role in thrombosis, GPVI is increasingly recognized to be critically involved in the maintenance of vascular integrity under conditions of inflammation, a process that is mechanistically distinct from normal hemostasis.23-26 As outlined above, the tumor environment is characterized by inflammation, indicating that platelet GPVI could have similar function in the cancer context. However, the role of platelet GPVI for the maintenance of vascular integrity in tumors has not been studied.
Using syngeneic mouse models of prostate and breast cancer, we show that the functional inhibition of GPVI rapidly induces profound tumor hemorrhage, diminishes tumor growth, and increases intratumoral accumulation of coadministered chemotherapeutic drugs, resulting in a markedly increased antitumor effect.
Materials and methods
General
If not otherwise indicated, chemicals were purchased from Sigma-Aldrich (Munich, Germany) or Carl Roth (Karlsruhe, Germany).
Animal experiments
All animal procedures described in this study were approved by the Regional Administration of Unterfranken (Lower District), Würzburg, Germany. The experiments were performed in accordance with relevant guidelines and regulations.
Cell lines and implantation
TrampC1 (prostate cancer) and AT-3 (breast cancer) cell lines were cultured at 37°C in a humidified atmosphere of 5% CO2 in Dulbecco’s modified Eagle medium supplemented with 10% fetal calf serum and 1% penicillin/streptomycin. For the heterotopic prostate cancer model, 5 × 106 TrampC1 cells in 50 µL phosphate-buffered saline were injected subcutaneously into the back of 8-week-old male C57BL/6J mice. A total of 0.5 × 106 AT-3 cells in 50 µL phosphate-buffered saline were injected dorsal of the inguinal mammary gland into the mammary fat pad of 8-week-old female virgin C57BL/6J mice. Tumor volumes were measured using Vernier calipers and determined using following calculation: volume = π/6 × l × w.2 In treatment, studies where tumor growth was a critical outcome assessment of tumor volume was performed blinded by a second experimenter.
Platelet depletion, blockade of GPVI, and neutrophil depletion
Platelets were depleted by IV injection of 50 µg of the αGPIb immunoglobulin G (IgG) R300 (Emfret Analytics, Eibelstadt, Germany) per mouse. To block GPVI function, 100 µg JAQ1 F(ab′)2 or soluble GPVI dimer27,28 (mGPVI-Fc, 4 mg/kg) was injected IV per mouse. Control animals were injected with 100 µg nonimmune rat IgG F(ab′)2. Neutrophils were depleted by intraperitoneal injection of 500 µg Ly6G antibody (RB6-8C5).14
Statistical analysis
Data are presented as mean ± standard deviation (SD; bar graphs) or mean ± standard error of the mean (SEM; growth curves) and were analyzed by the Kruskal-Wallis with Dunn’s multiple comparisons test or 2-way analysis of variance with Tukey’s multiple comparison test using Graph-Pad Prism 6 Software. P < .05 was considered as statistically significant.
Additional methods can be found in supplemental Methods (available on the Blood Web site).
Results
Genetic deficiency of GPVI leads to hemorrhages in tumors
GPVI deficient (Gp6−/−) mice on a C57BL/6 genetic background and wild-type littermates were implanted subcutaneously with TrampC1 cells (heterotopic prostate cancer model) or AT-3 cells into the mammary fat pad (orthotopic breast cancer model), and tumor size was evaluated every second day. TrampC1 tumors grew for 33 days to a volume of ∼250 mm3 (Figure 1A). AT-3 tumors grew for 21 days to a volume of 400 mm3 (Figure 1B). We found that genetic GPVI deficiency did not affect tumor growth. However, hemorrhage was observed in the tumors of Gp6−/− animals, reflected by a significant increase in hemoglobin content compared with controls (Figure 1C-E).
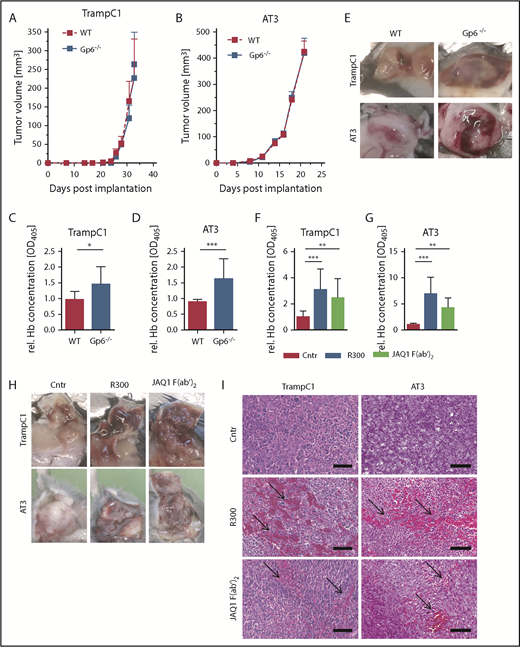
Increased bleeding in TrampC1 and AT-3 primary tumors in GPVI-deficient animals and upon GPVI blockade. TrampC1 prostate (A) and AT-3 breast cancer (B) tumors were grown in GPVI-deficient (Gp6−/−) mice. Tumor growth was measured every second day for 31 (A) or 21 (B) days postimplantation (n = 8; mean ± SEM). (C-E) The relative hemoglobin (rel. Hb) content was measured 31 or 21 days postimplantation (n = 8; mean ± SD). Effect of platelet depletion (R300 antibody against GPIbα) and GPVI blockade (JAQ1 F(ab′)2) on TrampC1 (F) and AT-3 (G) primary tumors grown in C57BL/6 animals 18 hours after antibody treatment. Relative quantification of the hemoglobin content in TrampC1 (F; n = 15) and AT-3 tumors (G; n = 10) (mean ± SD). Representative pictures (H) and hematoxylin and eosin staining (I) of the tumors. Arrows indicate accumulation of red blood cells in the tumor tissues. Scale bars, 100 µm. ***P < .001, **P < .01, and *P < .05; Kruskal-Wallis with Dunn’s multiple comparisons test. WT, wild-type.
Increased bleeding in TrampC1 and AT-3 primary tumors in GPVI-deficient animals and upon GPVI blockade. TrampC1 prostate (A) and AT-3 breast cancer (B) tumors were grown in GPVI-deficient (Gp6−/−) mice. Tumor growth was measured every second day for 31 (A) or 21 (B) days postimplantation (n = 8; mean ± SEM). (C-E) The relative hemoglobin (rel. Hb) content was measured 31 or 21 days postimplantation (n = 8; mean ± SD). Effect of platelet depletion (R300 antibody against GPIbα) and GPVI blockade (JAQ1 F(ab′)2) on TrampC1 (F) and AT-3 (G) primary tumors grown in C57BL/6 animals 18 hours after antibody treatment. Relative quantification of the hemoglobin content in TrampC1 (F; n = 15) and AT-3 tumors (G; n = 10) (mean ± SD). Representative pictures (H) and hematoxylin and eosin staining (I) of the tumors. Arrows indicate accumulation of red blood cells in the tumor tissues. Scale bars, 100 µm. ***P < .001, **P < .01, and *P < .05; Kruskal-Wallis with Dunn’s multiple comparisons test. WT, wild-type.
Functional inhibition of GPVI induces profound hemorrhage only at the tumor site
To further study the effect of pharmacological GPVI inhibition on intratumoral vascular integrity, wild-type C57BL/6 mice were implanted with TrampC1 or AT-3 cells. When tumors reached an average size of 300 mm3, the mice were divided into 3 groups in which (1) thrombocytopenia was induced using the R300 platelet depletion antibody, (2) GPVI was functionally inhibited with JAQ1 F(ab′)2, or (3) platelet function remained unaffected upon treatment with an irrelevant IgG F(ab′)2 control. Treatment with R300 led to an almost complete removal of platelets from the circulation within minutes, while injection of JAQ1 F(ab′)2 had no effect on platelet count compared with the IgG control in naive and tumor-bearing mice (supplemental Figure 1).29,30 In line with previous reports,31 we observed a drastic increase in intratumoral hemorrhage upon platelet depletion within 18 hours after treatment (Figure 1F-H). Of note, only a platelet count reduction to <5% of control induced significant hemorrhage in tumors (supplemental Figure 2). Remarkably, a comparable effect could be observed upon administration of JAQ1 F(ab′)2 with the relative hemoglobin concentration in tumors being 2 to 4 times higher compared with the IgG F(ab′)2-treated mice in both tested cancer models. Histopathological analysis revealed extravasated red blood cells in tumors isolated from mice after blockade of GPVI or depletion of platelets, whereas tumors from control IgG F(ab′)2 treated mice were unaffected (Figure 1I). No hemorrhage was observed in other organs such as spleen, liver, intestine, kidney, and lung of animals treated with IgG F(ab′)2 control, R300, or JAQ1 F(ab′)2 (supplemental Figure 3).
We also tested for potential off-target effects of JAQ1 F(ab′)2 in tumor-bearing Gp6−/− mice. The treatment had no detectable effects in these animals (ie, no inhibition of tumor growth or further increase in hemoglobin concentrations and hemorrhage), thereby confirming the specificity of JAQ1 F(ab′)2 in this experimental setting (supplemental Figure 4).
To characterize the damage of tumor vascular integrity induced by platelet depletion or GPVI inhibition, we performed coimmunofluorescent staining for the endothelial marker CD31 and the basement membrane components of tumor vessels, laminin α4 and collagen IV. Interestingly, the lumen size of the vessels was dramatically decreased in the animals treated with R300 or JAQ1 F(ab′)2 compared with controls, indicating vascular collapse (Figure 2A-B). However, the staining pattern of basement membrane components in noncollapsed vessels was comparable among all groups (Figure 2C-D). Altogether, these results show that pharmacological inhibition of GPVI rapidly destabilizes vascular integrity in tumors, leading to profound tumor hemorrhage, while not exerting such an effect in vessels of nontumor tissue.

Decreased proliferation and increased apoptosis in TrampC1 and AT-3 primary tumors upon GPVI blockade. (A) AT-3 primary tumors 18 hours after antibody treatment stained for CD31 (red), Col I (cyan), and 4′,6-diamidino-2-phenylindole (DAPI; blue). Scale bars, 100 µm. (B) For quantification of the amount of vessels with lumen, the average of 10 visual fields per mouse was calculated (n > 4). (C) AT-3 primary tumors 18 hours after antibody treatment stained for CD31 (red), Col IV (green), and DAPI (blue). Scale bars, 10 µm. (D) AT-3 primary tumors 18 hours after antibody treatment stained for CD31 (red), LMα4 (green), and DAPI (blue). Scale bars, 10 µm. Data are presented as mean ± SEM. (E) TrampC1 primary tumors 18 hours after antibody treatment stained for phospho-histone H3 (PH3) and cleaved caspase-3 (Cas3). Scale bars, 250 µm. (F) For quantification in the (left) TrampC1 model with n > 5 and (right) AT-3 model with n > 4, the average from 10 visual fields per mouse was calculated. ***P < .001, **P < .01, and *P < .05; Kruskal-Wallis with Dunn’s multiple comparisons test.
Decreased proliferation and increased apoptosis in TrampC1 and AT-3 primary tumors upon GPVI blockade. (A) AT-3 primary tumors 18 hours after antibody treatment stained for CD31 (red), Col I (cyan), and 4′,6-diamidino-2-phenylindole (DAPI; blue). Scale bars, 100 µm. (B) For quantification of the amount of vessels with lumen, the average of 10 visual fields per mouse was calculated (n > 4). (C) AT-3 primary tumors 18 hours after antibody treatment stained for CD31 (red), Col IV (green), and DAPI (blue). Scale bars, 10 µm. (D) AT-3 primary tumors 18 hours after antibody treatment stained for CD31 (red), LMα4 (green), and DAPI (blue). Scale bars, 10 µm. Data are presented as mean ± SEM. (E) TrampC1 primary tumors 18 hours after antibody treatment stained for phospho-histone H3 (PH3) and cleaved caspase-3 (Cas3). Scale bars, 250 µm. (F) For quantification in the (left) TrampC1 model with n > 5 and (right) AT-3 model with n > 4, the average from 10 visual fields per mouse was calculated. ***P < .001, **P < .01, and *P < .05; Kruskal-Wallis with Dunn’s multiple comparisons test.
Antibody-mediated inhibition of GPVI results in increased tumor cell death
To investigate whether the increased hemorrhage induced by GPVI inhibition affects tumor cell viability, immunofluorescence staining was performed 18 hours after anti-GPVI treatment. In both cancer models, the number of apoptotic cells was increased upon GPVI inhibition compared with control, as assessed by detection of cleaved caspase-3 (Figure 2E-F). Accordingly, the amount of phospho-histone H3–positive cells was significantly decreased in JAQ1 F(ab′)2-treated mice compared with controls (Figure 2E-F). We also noted slightly increased apoptosis in tumors isolated from Gp6−/− mice, which was not statistically significant (supplemental Figure 5).
Antibody-mediated inhibition of GPVI improves delivery of chemotherapeutic agents into the tumors
To investigate whether the improved uptake of chemotherapeutic agents into the tumor tissues can be induced by GPVI inhibition, TrampC1 and AT-3 tumor-bearing mice were treated with tritium-labeled paclitaxel (3H-PTX) and Doxil (liposomal doxorubicin), respectively, in combination with F(ab′)2 of JAQ1 or control IgG. Tumors and different organs such as heart, liver, spleen, kidney, intestine, and lung were removed after 2 or 24 hours. Doxil and 3H-PTX concentrations were determined in the indicated organs. Doxil and 3H-PTX were present at higher concentrations in TrampC1 tumors 2 hours after anti-GPVI treatment compared with the control group (Figure 3A-B). In AT-3 tumors, a tendency toward a higher concentration of the agents was observed. In contrast, no significant increase of Doxil was detected in any other organ (supplemental Figure 6). Furthermore, after 24 hours, the amount of chemotherapeutic agent in the tumors of the control group dramatically decreased compared with its value after 2 hours, whereas it remained high in platelet-depleted or GPVI-inhibited mice (Figure 3A-B). These results indicate that inhibition of GPVI profoundly perturbed vascular integrity specifically in tumors, thereby facilitating the delivery of chemotherapeutic drugs to the tumor site and improving their antitumor effects.
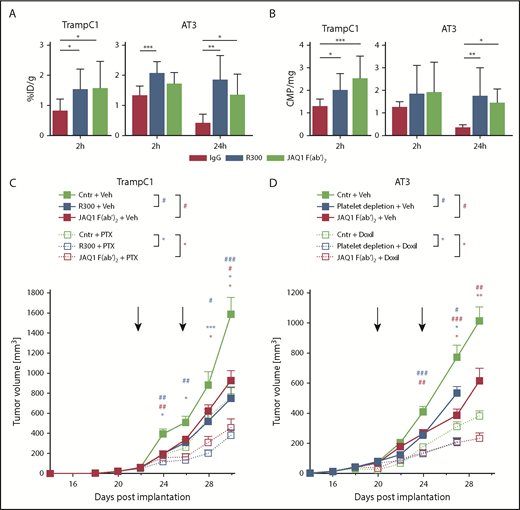
Combination of chemotherapeutic agents and GPVI blockade decreases tumor growth. TrampC1 and AT-3 tumor-bearing mice were treated with Doxil and 3H-PTX in combination with IgG control, R300, or JAQ1 F(ab′)2. After 2 and 24 hours, the concentration of Doxil (A, 2 hours) or 3H-PTX (B, 2 hours and 24 hours) within the tumor was measured. Mean with SD is displayed. ***P < .001, **P < .01, and *P < .05; Kruskal-Wallis with Dunn’s multiple comparisons test. Tumor volume was measured in TrampC1 (C) and AT-3 (D) tumor-bearing mice every second day. Data are presented as mean ± SEM. ***/###P < .001, **/##P < .01, and */#P < .05 (where * stands for the group treated with the chemotherapeutic agent and # the vehicle in the respective color); Kruskal-Wallis with Dunn’s multiple comparisons test. ID/g, injected dose per gram.
Combination of chemotherapeutic agents and GPVI blockade decreases tumor growth. TrampC1 and AT-3 tumor-bearing mice were treated with Doxil and 3H-PTX in combination with IgG control, R300, or JAQ1 F(ab′)2. After 2 and 24 hours, the concentration of Doxil (A, 2 hours) or 3H-PTX (B, 2 hours and 24 hours) within the tumor was measured. Mean with SD is displayed. ***P < .001, **P < .01, and *P < .05; Kruskal-Wallis with Dunn’s multiple comparisons test. Tumor volume was measured in TrampC1 (C) and AT-3 (D) tumor-bearing mice every second day. Data are presented as mean ± SEM. ***/###P < .001, **/##P < .01, and */#P < .05 (where * stands for the group treated with the chemotherapeutic agent and # the vehicle in the respective color); Kruskal-Wallis with Dunn’s multiple comparisons test. ID/g, injected dose per gram.
Decreased tumor growth upon GPVI inhibition in combination with chemotherapeutic agents
To investigate the effect of GPVI blockade on tumor growth in combination with chemotherapeutic drugs, TrampC1 and AT-3 tumor-bearing mice were treated with IgG control, R300, or JAQ1 F(ab′)2 as soon as the tumors were fully established (average size, >50 mm3). The treatment was repeated after 4 days. Tumor growth was significantly reduced in platelet-depleted and GPVI-inhibited mice compared with the control groups in both tested tumor models (Figure 3C-D). To analyze whether the tumor growth could further be diminished, the antiplatelet treatments were also combined in the same experiment with the injection of chemotherapeutic agents, paclitaxel (PTX) and Doxil, in TrampC1 and AT-3 tumor-bearing mice, respectively. Treatment of animals with the chemotherapeutic agents in combination with the control IgG decreased the tumor volume by 50% to 60%. Combination of these drugs with R300 or JAQ1 F(ab′)2 reduced tumor volumes by 70% to 80%, thus demonstrating improved efficacy of a combination treatment (Figure 3C-D). These results suggest that GPVI inhibition facilitates the delivery of Doxil and PTX to the tumor site and improves their antitumor effects.
Soluble GPVI dimer induces intratumor hemorrhage and inhibits tumor growth
To explore antitumor potential of an alternative, clinically relevant strategy interfering with GPVI function, we treated tumor-bearing mice with a soluble dimeric GPVI-Fc fusion protein,27 which competitively inhibits platelet adhesion on subendothelial collagens. Interestingly, we found that this treatment induced intratumoral hemorrhage and inhibited tumor growth to a similar extent as JAQ1 F(ab′)2 treatment (supplemental Figure 7). These results further confirmed the requirement of GPVI for the maintenance of tumor vascular integrity and suggest that blockade of this receptor by using soluble dimeric GPVI may represent an alternative strategy to inhibit tumor growth.
Neutrophil depletion reverts anti-GPVI–induced intratumoral bleeding and impaired tumor growth
Platelet depletion and anti-GPVI treatment caused bleeding in the tumor, but not in other organs. These results suggested that the abnormal vasculature in the tumor was more dependent on platelet GPVI function than the intact vasculature in other organs. Tumor-infiltrating white blood cells (WBCs), particularly neutrophils, have been described as a main source of proangiogenic factors but also major cause of tumor vessel damage and intratumoral hemorrhage.32-34 Indeed, we could observe major hemorrhagic spots in the tumors almost exclusively in areas of neutrophil accumulation (supplemental Figure 8). Therefore, we addressed whether the depletion of neutrophils may improve tumor vessel integrity, thereby affecting the bleeding phenotype in GPVI-inhibited mice. AT-3 tumor-bearing mice were treated with the RB6-8C5 antibody-depleting Ly6G+ cells 48 hours prior to treatment with IgG F(ab′)2, R300 or JAQ1 F(ab′)2. After 18 hours, the hemoglobin concentration in the tumors of these animals was determined (Figure 4A). Interestingly, Ly6G+ cell depletion by itself had no effect on intratumoral hemorrhage. Similar results were obtained with the TrampC1 model (supplemental Figure 9), indicating that neutrophils play a major role in the induction of hemorrhage in both cancer models. However, when mice were treated with JAQ1 F(ab′)2, Ly6G+ cell depletion protected the tumors from bleeding. The hemoglobin concentration of the thrombocytopenic mice with Ly6G+ cell depletion was significantly reduced compared with nontreated thrombocytopenic mice, but not to the level of control mice, indicating that also other factors in addition to Ly6G+ cells contribute to the induction of hemorrhage in thrombocytopenia.
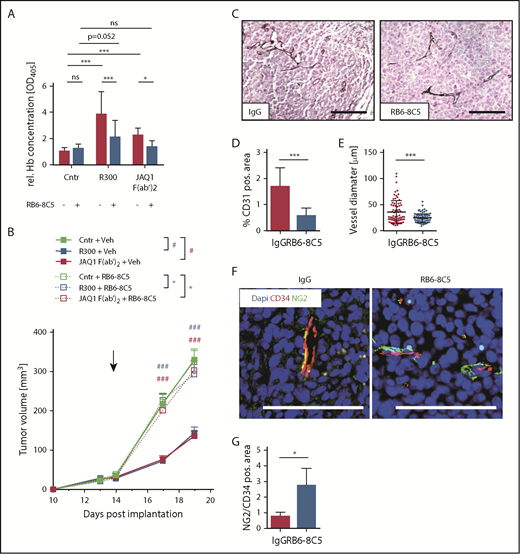
Vessel normalization by Ly6G+ WBC depletion with GPVI blockade reverts its effect on tumor growth. AT-3 tumor-bearing C57BL/6 mice were treated with RB6-8C5 antibody to deplete Ly6G+/Ly6C+ cells or with a vehicle control (saline) 48 hours before treatment with IgG control, R300, or the GPVI-blocking antibody. (A) After 18 hours, the relative hemoglobin content was measured (n ≥ 10; 2-way analysis of variance with Tukey’s multiple comparison test). (B) The tumor volume was measured every second day. Data are presented as mean ± SEM. ***/###P < .001, **/##P < .01, and */#P < .05 (where * stands for the group threated with the RB6-8C5 and # for the vehicle in the respective color); Kruskal-Wallis with Dunn’s multiple comparisons test. (C) Representative images of AT-3 tumor sections stained for CD31. Scale bars, 100 µm. (D) Vessel density after depletion of Ly6G+/Ly6C+ cells (n = 10; mean ± SEM). (E) Vessel diameter after depletion of Ly6G+/Ly6C+ cells. (F) Tumor sections stained for CD34 and the pericyte marker NG2. Scale bars, 100 µm. (G) Quantification of CD34+ and NG2+ areas in tumor sections after depletion of Ly6G+/Ly6C+ cells (n = 4; mean ± SD). ***P < .001, **P < .01, and *P < .05; Kruskal-Wallis with Dunn’s multiple comparisons test. ns, not significant.
Vessel normalization by Ly6G+ WBC depletion with GPVI blockade reverts its effect on tumor growth. AT-3 tumor-bearing C57BL/6 mice were treated with RB6-8C5 antibody to deplete Ly6G+/Ly6C+ cells or with a vehicle control (saline) 48 hours before treatment with IgG control, R300, or the GPVI-blocking antibody. (A) After 18 hours, the relative hemoglobin content was measured (n ≥ 10; 2-way analysis of variance with Tukey’s multiple comparison test). (B) The tumor volume was measured every second day. Data are presented as mean ± SEM. ***/###P < .001, **/##P < .01, and */#P < .05 (where * stands for the group threated with the RB6-8C5 and # for the vehicle in the respective color); Kruskal-Wallis with Dunn’s multiple comparisons test. (C) Representative images of AT-3 tumor sections stained for CD31. Scale bars, 100 µm. (D) Vessel density after depletion of Ly6G+/Ly6C+ cells (n = 10; mean ± SEM). (E) Vessel diameter after depletion of Ly6G+/Ly6C+ cells. (F) Tumor sections stained for CD34 and the pericyte marker NG2. Scale bars, 100 µm. (G) Quantification of CD34+ and NG2+ areas in tumor sections after depletion of Ly6G+/Ly6C+ cells (n = 4; mean ± SD). ***P < .001, **P < .01, and *P < .05; Kruskal-Wallis with Dunn’s multiple comparisons test. ns, not significant.
To test whether this also affected the tumor growth-inhibiting effect of platelet depletion and GPVI inhibition, we treated tumor-bearing mice with a combination of RB6-8C5 and R300 or JAQ1 F(ab′)2 (every 4 days). Depletion of Ly6G+ cells by itself did not affect tumor growth, as demonstrated previously in other models.35 In line with the absent bleeding, tumor growth was not reduced in Ly6G+ WBC-depleted animals upon platelet depletion or GPVI inhibition (Figure 4B).
Finally, we assessed whether depletion of Ly6G+ cells led to a stabilization of the tumor vessel wall. Indeed, treatment with RB6-8C5 decreased tumor vessel density and reduced the abnormally enlarged diameter of vessels in AT-3 tumors (Figure 4C-E). Moreover, Ly6G+-cell depletion also increased the coverage of blood vessels with NG2+ pericytes, demonstrating improved vessel stabilization and maturation (Figure 4F-G). Given this strong effect of Ly6G+ cells on tumor vessel stabilization and the documented role of platelets in fostering neutrophil infiltration, we wanted to exclude that the results observed so far were caused by effects of GPVI inhibition on neutrophil infiltration. Flow cytometric analysis showed comparable frequencies of Ly6G+ intratumoral neutrophils in mice treated with control F(ab′)2 and JAQ1 F(ab′)2 (supplemental Figure 10), suggesting that their recruitment was not dependent on GPVI. Thus, the relative specificity of platelet depletion and GPVI targeting for causing hemorrhage solely in the tumor appears to be based on defects in the tumor vasculature.
Discussion
In this study, we establish the platelet-specific receptor GPVI as a key regulator of vascular integrity in solid tumors and show that its inhibition may provide a new strategy to increase efficacy of antitumor chemotherapy.
Systemic therapeutics are a mainstay of cancer therapy and especially an indispensable instrument in the management of progressed, metastatic disease. The effectiveness of treatment with systemically applied therapeutics depends not only on the efficacy of the drug’s delivery into the targeted organs but also on whether these drugs can reach tumor cells in concentrations sufficient to exert their therapeutic effect. However, both the defectiveness of the vasculature and the abundant highly condensed extracellular matrix including collagens found in solid tumors represent physical barriers impeding effective drug transport into the tumor.36,37 Improving vascular function has long been discussed as a possibility to improve tumor drug transport.7 Inhibition of the pathological aspects of tumor angiogenesis by various approaches can increase accumulation and efficacy of tumor-directed systemic drugs.38-41 Following this idea, we demonstrate here that exploiting the vulnerability of the already damaged tumor vasculature by provoking a complete vascular collapse by platelet targeting can facilitate drug delivery to tumor sites.
Platelets have been demonstrated to be critical for maintaining a minimum of tumor vascular integrity. Ho-Tin-Noé et al have shown that platelet depletion selectively renders tumor vessels highly permeable while leaving vessels in other organs unaffected.31,42 Another previous study showed that the induction of acute thrombocytopenia resulted in decreased tumor growth in a mouse model of ovarian carcinoma.43 Furthermore, Demers et al also demonstrated that platelet depletion favored intratumoral accumulation of PTX, thereby enhancing its antitumor effects.15 We could reproduce this drug accumulation effect by inhibition of GPVI. PTX and liposomal Doxil accumulated much longer in treated tumors, and the effect of the safer treatment option with GPVI-inhibition was comparable to that of platelet depletion. In addition, we could demonstrate that the prolonged accumulation was caused by a flush-out-and-trapping effect (supplemental Figure 11). Although drug levels were already high immediately after coadministration of platelet-targeted antibodies and cytotoxic drug, the difference in drug concentration increased compared with control-treated mice. In the control animals, drug levels decreased significantly over the next 24 hours as the agents were secreted. In the R300 or JAQ1 F(ab′)2-treated tumors, drug levels did not change significantly and remained high during the observation period, as this compartment was secluded from the bloodstream after treatment-induced vascular collapse.
In contrast to some previous reports, we found that platelet depletion and GPVI inhibition had a profound effect on tumor growth. In line with the previous work, we observed strong hemorrhage and subsequent necrosis in the treated tumors, but this resulted in a reduction of tumor growth only in our models. In contrast, the B16 melanoma and 4T1 breast cancer models used by Ho-Tin-Noé et al were interestingly unaffected by these detrimental events.31,42 It could be possible that in these models oncogenic factors are different which may improve vascularization within the developing tumor, but this clearly requires further investigation.
Although R300 or JAQ1 F(ab′)2 treatment lead to an acute drastic undersupply resulting in large parts of up to 80% of the tumors undergoing necrosis, a complete stop of tumor growth or a regression could not be reached, even with repeated treatments. This is in line with previous observations after antiangiogenic therapy or treatment with vascular disrupting agents.44,45 These vascular intervention therapies mostly affect highly disorganized vessels in the center of the tumor while leaving the margin of the tumor that is supplied by more functional vessels unharmed. From this “viable rim,” the tumors can recuperate.
Our results revealed a key role of GPVI in safeguarding vascular integrity in growing tumors. Besides its critical function in thrombus formation, GPVI is increasingly recognized as a central modulator of thromboinflammatory pathologies.23 In an experimental glomerulonephritis model, GPVI supports platelet adhesion and leukocyte infiltration into the inflamed tissue.46,47 Furthermore, in a model of autoimmune rheumatoid arthritis, GPVI was shown to mediate the recruitment of platelets to inflamed joints, thereby increasing permeability of the synovial microvasculature and local recruitment of neutrophils.25 Although it may seem counterintuitive, as GPVI promotes opening of endothelial junctions and vascular permeability, in distinct inflammatory conditions, single platelets were shown to seal neutrophil-induced vascular breaches via GPVI.23 This was notably observed in a cutaneous reverse passive Arthus inflammation model, in which genetic deficiency or blockade of GPVI signaling increased bleeding.23 Interestingly, Gp6−/− mice challenged in the reverse passive Arthus model or an lipopolysaccharide-induced lung injury model did not display any bleeding phenotype.48 Altogether, these studies suggested that the role of GPVI in maintenance of inflammatory hemostasis is organ and stimulus dependent.
During cancer progression, innate immune cells (particularly neutrophils) migrate to tumor niches, thereby causing intratumoral hemorrhages and amplifying inflammatory reactions, resulting in more aberrant angiogenic signaling and promotion of the defective tumor vessel phenotype.32-34,49 Our findings also provide evidence that the platelet response required to prevent hemorrhage may be similar for cutaneous and tumor inflammation contexts. Bleeding was observed upon platelet depletion also in several other tumor types, such as subcutaneous Lewis lung carcinoma and B16F10 melanoma models,31 suggesting that targeting GPVI to improve the efficiency of drug delivery and chemotherapy could be feasible in a large variety of tumors.
We found that the vascular-disintegrating effect of GPVI inhibition was strongly diminished upon depletion of neutrophils. In line with this, previous studies showed that induction of thrombocytopenia in β2 and β3 integrin–deficient mice, which are characterized by a reduction of infiltrating macrophages and neutrophils in the tumor, resulted in decreased tumor hemorrhage compared with control mice with thrombocytopenia.42
Our results suggest that in the here used tumor models neutrophils are to a large extent responsible for the bleeding associated with thrombocytopenia or GPVI inhibition. Since neutrophil recruitment into the tumors was not GPVI dependent (supplemental Figure 10), it appears that GPVI is required to “repair” or limit neutrophil-induced vascular damage. Indeed, we found major hemorrhagic spots in the tumors almost exclusively at sites of neutrophil accumulation (supplemental Figure 8). However, for a detailed understanding of the exact mechanisms leading to intratumor bleeding and its prevention by platelets, dynamic imaging of neutrophil-endothelial cell-platelet interactions in the tumor will be required.
Obviously, neutrophils fulfill overall the same functions in innate immunity in humans and mice. However, there are also potentially relevant species differences, especially with respect to relative leukocyte numbers, certain subpopulations (N1/N2), or attributed granule functions (ie, defensin expression),50 making a direct extrapolation of mouse studies to human patients difficult.
Neutrophils are rich sources of reactive oxygen species, generating enzymes, proteases, and matrix metalloproteases,49 and platelets have been reported to inhibit (or uptake and store) cytotoxic releasate from tumor-inflicting neutrophils, which might contribute to their vessel-protective effect.51-53 A possible role of GPVI in this process has not been studied so far.
Another possible mechanism was recently proposed by Gros et al,23 who demonstrated in a model of skin inflammation that single platelets seal neutrophil-induced vascular leakage in a GPVI-dependent manner, indicating that they do so by attaching to exposed subendothelial collagens and possibly laminins. This idea is supported by our results showing that a soluble GPVI dimer,27 which competitively inhibits platelet adhesion on exposed collagen matrix, induces intratumoral hemorrhage and reduces tumor growth to a similar extent as JAQ1 F(ab′)2 treatment (supplemental Figure 7).
Another important finding of our study is that pharmacological GPVI inhibition produced a more pronounced tumor hemorrhage than genetic GPVI deficiency. The observation that the sudden blockade of a platelet adhesion receptor may have stronger effects than its genetic deficiency may not be entirely unexpected, as this has previously also been reported for GPIIb/IIIa in the setting of ischemic stroke. While GPIIb/IIIa inhibitors provoke massive intracranial hemorrhage in acute stroke in mice54 and humans,55,56 no such effect is seen in mice genetically deficient for GPIIb(IIIa).57 Although the exact underlying pathways remain to be identified, this strongly suggests that compensatory mechanisms are activated in affected mice (and probably also in humans) to minimize hemostatic and/or vascular defects resulting from the genetic deficiency in platelet effector functions.
We showed that antibody-mediated blockade of GPVI or induction of thrombocytopenia enhanced the accumulation of two different cytotoxic drugs, Doxil and PTX, specifically in tumors. There are also indications that platelets directly increase resistance against PTX.58 Platelets increased survival of tumor cells in culture subjected to PTX, even if coincubated without direct contact to the tumor cells. This suggests that platelet secreted factors might have chemoprotective effects, at least against PTX.
Earlier, other platelet adhesion receptors such as GPIIb/IIIa and GPIb have also been proposed as antitumor targets as they have been implicated in tumor growth and metastasis.59-64 While these receptors are essential for normal hemostasis, their genetic deficiency did not cause major inflammation-induced bleeding in most mouse models in different organ systems (reviewed in Bergmeier and Stefanini65 and Rayes et al66 ). Future studies are needed to address their role in the maintenance of vascular integrity in different tumor models.
Platelets contribute to tumor metastasis, eg, by shielding circulating tumor cells from the immune system in the blood and actively enhancing tumor cell migration and extravasation.59,64,67-69 Earlier, the potential of GPVI inhibition to reduce the capacity for metastasis formation was demonstrated.70 This indicates multiple beneficial effects of GPVI inhibition by concomitantly decreasing the primary tumor growth, increasing drug deposition, reducing chemoprotection, and inhibiting further metastasis. Of note, GPVI is exclusively expressed on megakaryocyte/platelet lineage, which largely excludes risk of side effects of anti-GPVI agents on other cell types. Due to its role in thrombotic diseases, a considerable effort is currently put into developing GPVI inhibitors. Recently, the soluble dimeric GPVI receptor fusion protein, Revacept, has been successfully evaluated in a phase 1 clinical trial and is currently undergoing a phase 2 trial for the treatment of a variety of thrombotic pathologies.22,71 In light of these observations and our results, targeting GPVI may represent a seducing approach associating safety to efficacy not only toward thrombotic diseases but also beyond.
In conclusion, our studies highlighted a crucial tumor-supporting role of GPVI and also provide a proof of concept that targeting of this platelet receptor could be therapeutically effective against cancers.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Ulrich E. Schaible (Research Center Borstel) for kindly providing the RB6-8C5 hybridoma, Andreas Friebe (Institute of Physiology, University of Würzburg) for help with scintillation counting, and Ivana Jorgacevic (Institute of Experimental Biomedicine, University Hospital Würzburg) for help with flow cytometry experiments.
This work was supported by the Deutsche Forschungsgemeinschaft (SFB/TR 240) (B.N.). J.V. was supported by a grant of the German Excellence Initiative to the Graduate School of Life Sciences, University of Würzburg. E.M.B. was supported by Europäischer Fonds für regionale Entwicklung-Bayern.
Authorship
Contribution: J.V., E.M.-B., J.G.-P., and R.N. acquired the data; K.R., A.Z., and S.E. interpreted the data; L.S. and S.I.A. provided essential tools and interpreted the data; E.H. and B.N. designed the research; and J.V., E.M.-B., E.H., and B.N. analyzed the data and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Erik Henke, Institute of Anatomy and Cell Biology, University of Würzburg, Koellikerstraße 6, 97070 Würzburg, Germany; e-mail: erik.henke@uni-wuerzburg.de; and Bernhard Nieswandt, Institute of Experimental Biomedicine, University Hospital and Rudolf Virchow Center, University of Würzburg, Josef-Schneider-Straße 2, 97080 Würzburg, Germany; e-mail: bernhard.nieswandt@virchow.uni-wuerzburg.de.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal