

Key Points
Human neutrophils express low levels of FcγRIIIA, which are usually masked by the high levels of homologous FcγRIIIB.
FcγRIIIA, but not FcγRIIIB, appears to mediate neutrophil activation by IgG antibodies, as well as phagocytosis of antibody-opsonized beads.

Abstract
We have identified a rare healthy FcγRIIIB (CD16B)-null donor completely lacking FCGR3B RNA and protein expression and dissected the role of the different neutrophil Fcγ receptors in the response to therapeutic anti-CD20 monoclonal antibodies. We observed that polymorphonuclear neutrophils (PMNs) from FcγRIIIB wild-type (WT) individuals or the null donor were more effectively activated by chronic lymphocytic leukemia (CLL) B-cell targets opsonized with glycoengineered anti-CD20 antibodies compared with fully core-fucosylated anti-CD20 antibodies, suggesting the presence and role of FcγRIIIA (CD16A) on PMNs. Indeed, we demonstrated by reverse-transcription polymerase chain reaction, flow cytometry, and western blot analysis that PMNs from FcγRIIIB WT donors and the null individual express low levels of FcγRIIIA on their surfaces. FcγRIIIA is a functional and activating molecule on these cells, because anti-CD16 F(ab′)2 antibodies alone were able to activate highly purified PMNs from the FcγRIIIB-null donor. Use of blocking anti-CD16 and anti-CD32 antibodies showed that FcγRIIIA is also a major mediator of phagocytosis of CD20-opsonized beads by FcγRIIIB WT and null PMNs. In contrast, trogocytosis of antibody-opsonized CLL B cells by PMNs was mediated primarily by FcγRIIIB in WT PMNs and by FcγRIIA in null PMNs. We conclude that FcγRIIIA is an important player in PMN functions, whereas FcγRIIIB is dispensable for activation and phagocytosis. We discuss the clinical implications of these findings.
Introduction
Unconjugated immunoglobulin G1 (IgG1) therapeutic antibodies, such as anti-CD20, act in vivo primarily through Fcγ receptors (FcγRs) (FcγRIII [CD16], FcγRII [CD32], and FcγRI [CD64]) and immune-mediated mechanisms, including antibody-dependent cellular cytotoxicity (ADCC) by natural killer (NK) cells and phagocytosis by macrophages.1 Polymorphonuclear neutrophils (PMNs) also appear to be important, because PMN depletion in mice reduced or abolished the therapeutic activity of anti-CD20 and other monoclonal antibodies (MAbs).2-5 In vitro, anti-CD20 and anti-EGFR MAbs activate PMNs and induce production of reactive oxygen species.6,7 Anti-EGFR MAbs, but not anti-CD20 MAbs, also induce ADCC by PMNs.6,8-10 More recently, we have demonstrated that human PMNs mediate trogocytosis, rather than phagocytosis, of chronic lymphocytic leukemia (CLL) B cells opsonized with anti-CD20 antibodies, although they are able to phagocytose smaller particles coated with antibodies.11
Low-fucose–containing therapeutic antibodies (eg, anti-CD20 obinutuzumab [OBZ]) are known to bind more efficiently than fully core-fucosylated MAbs to FcγRIIIA and FcγRIIIB as a result of the high degree of homology (>95%) between the extracellular portions of these 2 FcγRs. On the other hand, fucose content does not affect binding to the other FcγRs: FcγRI and FcγRIIA/B.6,12-15 Glycoengineered anti-CD20 antibodies have been shown to activate neutrophils more efficiently than the unmodified versions of the same antibodies, suggesting that FcγRIIIB plays an important role in neutrophil activation.6 Indeed, PMNs express high levels of FcγRIIIB and FcγRIIA, and both of these FcγRs have been implicated in antibody-mediated PMN activation.16 FcγRI is expressed weakly on resting neutrophils but is induced only after PMN activation, in particular following stimulation with IFN-γ.17
FcγRIIIA and FcγRIIIB are encoded by different genes (FCGR3A and FCGR3B, respectively) that have arisen by duplication during evolution, and both are polymorphic molecules.16,18,19 FcγRIIIA associates with ITAM-containing signaling molecules (FcRγ or CD3ζ chain homodimers or heterodimers), and this interaction is required for FcγRIIIA surface expression and signaling.20-23 Immune complex (IC) binding to the FcγRIIIA complex leads to Ca2+ flux, phosphatidyl inositol 4,5 biphosphate hydrolysis, and an intracellular phosphorylation cascade.23-25 In contrast, FcγRIIIB is glycosylphosphatidylinositol linked, does not interact with FcRγ or CD3ζ, and has no signaling capacity by itself.16,23,25 FcγRIIIB has been suggested by some investigators to activate PMN functions through interaction with other molecules.26
The most common variants of FcγRIIIB are FcγRIIIB-HNA1a and FcγRIIIB-HNA1b (previously known as NA1 and NA2), with a frequency of 0.30 to 0.68 and 0.30 to 0.70, respectively, according to ethnic group.27 Other rare polymorphisms exist, in particular FcγRIIIB-SH (frequency of 0.05 in whites) and FcγRIIIB null, showing deletion of FCGR3B and part of the FCGR2C genes (frequency ∼ 0.03-0.1).28,29 No murine equivalent of FcγRIIIB/FCGR3B exists.16
We have identified a very rare FcγRIIIB-null healthy individual and, using this donor, dissected the role of FcγRIIIB and FcγRIIIA in PMN activation by IgG1 antibodies and other neutrophil functions.
Materials and methods
Antibodies
OBZ (GA101), rituximab (RTX), control irrelevant anti-EGFR cetuximab (CTX; ERBITUX; Bristol-Myers-Squibb), and ofatumumab (OFA) were obtained from a local pharmacy. Glycoengineered RTX (RTX-GE) was a kind gift of C. Klein (Roche Glycart, Schlieren, Switzerland). For blocking experiments, anti-CD16 (clone 3G8, mIgG1) and anti-CD32 (clone 7.3, mIgG1) F(ab′)2 fragments (Ancell, Bayport, MN) or whole IgGs were used (BD Biosciences, San Jose, CA for anti-CD16 3G8 and Ancell for anti-CD32 7.3 antibody). For western blotting, anti-CD16A/B (GRM1, mIgG2a; Santa Cruz Biotechnology, Dallas, TX) and polyclonal anti-FcγRIIIA/CD16A specific intracellular peptide antibody (Atlas Antibodies, Bromma, Sweden), as well as anti-actin antibody (Santa Cruz Biotechnology), were used. For flow cytometry, anti-CD32–FITC and anti-CD64–FITC (both from BD Biosciences), anti-CD16–FITC (MEM-154, mIgG1; Immunological Sciences and 3G8; BD Biosciences), and unlabeled anti-CD16 (clone CLB-gran/11, mIgG2; Abcam, Cambridge, UK, and clone GRM1; Santa Cruz Biotechnology) antibodies were used. For unlabeled primary antibodies, goat anti-mouse IgG–FITC antibody (BD Biosciences) was used for detection.
Cells
Blood in desirudin or EDTA was obtained from healthy volunteers or untreated CLL patients after informed consent. The study was approved by the hospital’s Ethical Committee. For whole-blood assays, peripheral blood was drawn in 50 µg/mL desirudin (IPRIVASK; Canyon Pharmaceuticals, Basel, Switzerland).30 PMNs were generally purified to ≥95% purity from peripheral blood by negative selection using a MACSxpress Neutrophil Isolation Kit, according to the manufacturer’s instructions (Miltenyi Biotec, Bergisch Gladbach, Germany). For reverse-transcription polymerase chain reaction (RT-PCR) experiments, the mononucleated cell (MNC) and granulocyte fractions were separated by standard Ficoll-Hypaque gradient centrifugation (Seromed, Berlin, Germany), followed by purification with a MACSxpress Neutrophil Isolation Kit to obtain >99.5% pure PMNs. Monocytes were purified from the MNC fraction using CD14 MicroBeads and LS columns for positive selection of cells (Miltenyi Biotec). The CLL cells used were peripheral blood MNCs from CLL patients that contained >90% CLL B cells and that had been aliquoted and frozen in nitrogen vapors.
PMN activation
Either 100 µL of whole blood in desirudin or purified PMNs (5 × 105 per well) were plated with peripheral MNCs from CLL patients at a 1:3 PMN/CLL cell ratio, opsonized with 0.001 to 10 µg/mL anti-CD20 antibody. In some cases, 10 µg/mL anti-CD16 (clone 3G8) and anti-CD32 (clone 7.3) F(ab′)2 or whole IgG was added instead of opsonized CLL cells. Samples were incubated for 4 hours at 37°C, 5% CO2 and then stained for 20 minutes with anti-CD11b–PE antibody (clone D12) or CD62L-APC (both from BD Biosciences, Franklin Lakes, NJ) to measure PMN activation. In some cases, FITC-labeled CBRM1/5 clone (Santa Cruz Biotechnology) was used to specifically detect activated CD11b (neoantigen). Whole-blood samples were then lysed with hypotonic lysis solution (Pharm Lyse; BD Biosciences) and analyzed on a FACSCanto II instrument (BD Biosciences).
Phagocytosis of opsonized beads
Because PMNs do not phagocytose CLL B cells efficiently,11 we used 5-µm polystyrene microparticles (Sigma, St. Louis, MO) as targets. Washed sterile beads were coated by incubation for 1 hour at room temperature with 6.7 mM RTX or fibronectin (Sigma) in Hank’s balanced salt solution. Beads were then washed 3 times before use and counted. Purified PMNs were incubated with 10 µg/mL blocking anti-FcγRIII/II F(ab′)2 for 5 minutes before adding opsonized beads at a 1:10 ratio (PMNs/beads) in 8-well Lab-Tek Chamber Slides (Nunc, Rochester, NY). After incubation for 1 hour at 37°C, the slides were washed gently in PBS and fixed in 2% paraformaldehyde. Four random fields were photographed for each sample, and the number of phagocytosing cells and number of beads per 100 cells were counted in a blinded fashion. At least 200 PMNs were counted for each condition. Percentage phagocytosis was defined as the number of PMNs having phagocytosed ≥1 bead × 100/total number of PMNs.
Trogocytosis of opsonized CLL B cells
Trogocytosis experiments were performed as previously described.11 Briefly, purified unlabeled PMNs were incubated for 4 hours at 37°C, 5% CO2 with PKH67-labeled CLL B cells at a 1:3 effector-to-target ratio in the presence of 1 µg/mL anti-CD20 antibodies. In some cases, the purified PMNs were incubated for 5 minutes with 10 µg/mL anti-CD16/32 blocking IgGs before adding them to the coculture. Transfer of PKH67 to PMNs was analyzed by flow cytometry on a FACSCanto II instrument (BD Biosciences), identifying PMNs by their scatter properties.
Western blots
Cells were counted and lysed in 2× Laemmli reducing sample buffer (Sigma). Lysates from 2.5 × 105 cells were loaded in each lane in standard polyacrylamide gels. Blocking and antibody binding were performed in Tris-buffered saline-Tween buffer containing 0.5% nonfat skimmed milk. Detection was performed using the relative horseradish peroxidase–labeled secondary antibody (Santa Cruz Biotechnology) and Super Signal West Pico or Dura Chemiluminescent Substrate (Pierce, Rockford, IL).
FCGR3B genotyping
Genomic DNA was isolated from MNCs using a Gentra Puregene Blood kit (QIAGEN, Germantown, MD). CD16B genotyping was performed as described,31 using 50 ng of genomic DNA, specific FCGR3B-HNA1a and FCGR3B-HNA1b forward primers, and 1 common reverse primer (supplemental Table 1, available on the Blood Web site). Thirty-five amplification cycles were performed using a 9800 Applied Biosystems Instrument (Thermo Fisher Scientific, Waltham, MA) with 55°C annealing temperature. The 141-bp (HNA1a) and 219-bp (HNA1b) amplification products were separated by standard agarose gel electrophoresis. As control, the Jak2 gene was amplified, yielding the expected 364-bp band.
FCGR3A and FCGR3B expression by RT-PCR
Total RNA was extracted by standard procedure using TRIzol Reagent (Invitrogen, Carlsbad, CA), chloroform, and isopropanol precipitation. Reverse transcription was performed using SuperScript II Reverse Transcriptase (Invitrogen-Thermo Scientific, Waltham, MA), according to standard procedures. For amplification, AmpliTaq Gold (Applied Biosystems-Thermo Scientific, Waltham, MA) and 2 sets of primers specific for CD16A and CD16B were used, within exon 5 or across exons 3 and 4 (supplemental Table 1), and MgCl2 concentrations were optimized for each set (1.5 and 2.0 mM, respectively). Polymerase chain reaction (PCR) products were cut with TaqI enzyme for 1 hour at 65°C, and bands were resolved in 4% agarose gels. PCR followed by TaqI digestion was expected to yield products of 118 + 59 nucleotides (nt)(FCGR3A) or 177 nt (FCGR3B) in case of exon 5 primers, and 219 + 101 nt (FCGR3A) or 246 + 122 nt (FCGR3B) in case of exon 3 to 4 primers.
Statistical analysis
The data were analyzed using a paired or unpaired Student t test, as appropriate.
Results
Identification of an FcγRIIIB-null healthy donor
To dissect the role of FcγRIIIB in PMN activation by therapeutic antibodies, we initially performed PCR analysis of 25 donors to determine the FCGR3B-HNA1a (NA1) and FCGR3B-HNA1b (NA2) allotypes of healthy donors in our institution. These allotypes were identified with the expected frequency.27,28 During analysis, we identified a healthy donor (#12) whose DNA did not amplify with the primers specific for HNA1a and HNA1b (Figure 1A). The SH allele is a much rarer polymorphic form of CD16B but would have been amplified by the HNA1b primers used.27 Therefore, the data suggested that donor 12 was a null donor for FCGR3B/FcγRIIIB.28,29
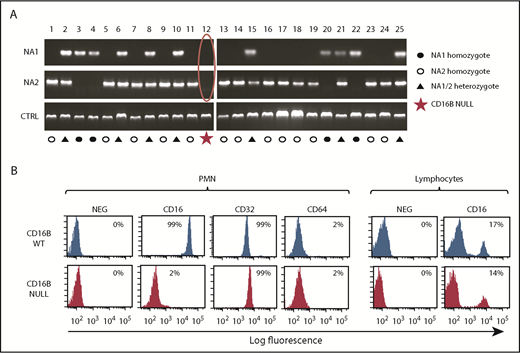
Identification of a rare FcγRIIIB-null donor lacking FCGR3B gene and protein. (A) Genomic PCR was performed on DNA from 25 healthy donors, using primers specific for the HNA1a (NA1) and HNA1b (NA2) allotypes. Jak2 gene was amplified as control. (B) Whole blood from FcγRIIIB WT donors and the FcγRIIIB-null donor was stained with FITC-labeled anti-CD16, anti-CD32, and anti-CD64 antibodies and selected populations, based on scatter properties corresponding to granulocyte and lymphocytes, analyzed by flow cytometry. The data shown are representative of ≥3 experiments.
Identification of a rare FcγRIIIB-null donor lacking FCGR3B gene and protein. (A) Genomic PCR was performed on DNA from 25 healthy donors, using primers specific for the HNA1a (NA1) and HNA1b (NA2) allotypes. Jak2 gene was amplified as control. (B) Whole blood from FcγRIIIB WT donors and the FcγRIIIB-null donor was stained with FITC-labeled anti-CD16, anti-CD32, and anti-CD64 antibodies and selected populations, based on scatter properties corresponding to granulocyte and lymphocytes, analyzed by flow cytometry. The data shown are representative of ≥3 experiments.
To confirm that donor 12 specifically lacks FcγRIIIB protein expression, we performed a phenotypic analysis of his peripheral blood cells using a pan-FcγRIIIA/B MAb. Whereas the antibody strongly stained the PMNs of the FcγRIIIB wild-type (WT) donors (99%; mean fluorescence intensity [MFI], 34 307), no significant staining could be observed in PMNs from the null donor (2%; MFI, 257) (Figure 1B). In contrast, the resting PMNs of FcγRIIIB WT and null donors stained strongly with FcγRII (CD32) antibodies, but not with FcγRI (CD64) antibodies, as expected (Figure 1B). We could also detect FcγRIII in ∼15% of lymphocytes from all donors, including those of donor 12, presumably detecting FcγRIIIA on NK cells, as expected (Figure 1B).29
Glycoengineered anti-CD20 antibodies activate PMNs more efficiently whether they derive from FcγRIIIB WT donors or the null individual
Having identified a donor fully lacking FcγRIIIB expression, we went on to investigate the functional role of this molecule in PMN activation by therapeutic anti-CD20 antibodies, using nonglycoengineered RTX and OFA, as well as RTX-GE and OBZ; the last 2 are known to bind more strongly to FcγRIIIA and FcγRIIIB.6,14,15 Activation was first performed in whole-blood assays and was measured by an increase in total CD11b and a decrease in CD62L expression at 6 hours by flow cytometry, as described previously.6 As shown in Figure 2A and 2B, glycoengineered CD20 antibodies (RTX-GE and OBZ) activated PMNs in whole-blood assays to the same extent, whether samples were derived from FcγRIIIB WT donors or the null individual. In contrast and as previously observed, nonglycoengineered OFA and RTX did not significantly activate PMNs in whole blood.6 These data suggested that glycoengineered anti-CD20 antibodies can activate PMNs more effectively than unmodified MAbs, even in the absence of FcγRIIIB; this was an unexpected result, because glycoengineering specifically affects FcγRIIIA/B binding.

PMNs from FcγRIIIB WT and null donors are activated more effectively by glycoengineered CD20 antibodies compared with unmodified CD20 antibodies. (A-B) Whole blood from FcγRIIIB donors or the FcγRIIIB-null individual was drawn in desirudin, and 10 µg/mL of the indicated anti-CD20 antibodies or control CTX (CTRL) was added. After 4 hours, PMN activation was measured by staining with anti-CD11b–PE (A) and anti-CD62L–APC (B) antibodies and flow cytometry. The results are the MFI of CD11b and CD62L expression on PMNs relative to irrelevant CTX-treated controls. (C-D) PMNs were purified from peripheral blood of FcγRIIIB WT and null donors and stimulated for 4 hours with CLL B cells opsonized with 10 µg/mL of the indicated CD20 antibodies or CTX (CTRL). PMN activation was measured by staining with anti-CD11b–PE (C) and anti-CD62L–APC (D) antibodies and flow cytometry. The results are the MFI of CD11b and CD62L expression on PMNs. All data are the means and standard deviations of 3 or 4 independent experiments. *P < .05, **P < .01, ***P < .001 vs the respective controls without antibody (CTRL).
PMNs from FcγRIIIB WT and null donors are activated more effectively by glycoengineered CD20 antibodies compared with unmodified CD20 antibodies. (A-B) Whole blood from FcγRIIIB donors or the FcγRIIIB-null individual was drawn in desirudin, and 10 µg/mL of the indicated anti-CD20 antibodies or control CTX (CTRL) was added. After 4 hours, PMN activation was measured by staining with anti-CD11b–PE (A) and anti-CD62L–APC (B) antibodies and flow cytometry. The results are the MFI of CD11b and CD62L expression on PMNs relative to irrelevant CTX-treated controls. (C-D) PMNs were purified from peripheral blood of FcγRIIIB WT and null donors and stimulated for 4 hours with CLL B cells opsonized with 10 µg/mL of the indicated CD20 antibodies or CTX (CTRL). PMN activation was measured by staining with anti-CD11b–PE (C) and anti-CD62L–APC (D) antibodies and flow cytometry. The results are the MFI of CD11b and CD62L expression on PMNs. All data are the means and standard deviations of 3 or 4 independent experiments. *P < .05, **P < .01, ***P < .001 vs the respective controls without antibody (CTRL).
To exclude a role for NK cells or monocytes in whole blood, we repeated the experiments using highly purified PMNs (>98%) stimulated with CLL B cells opsonized with RTX-GE or WT RTX. Also, in this case, the glycoengineered antibody induced significantly greater activation than the WT antibody using PMNs from FcγRIIIB WT donors or the null donor (Figure 2C-D), in agreement with the whole-blood assays (Figure 2A-B).
The results were also confirmed in dose-response curves: PMN activation by RTX-GE was similar whether these cells derived from FcγRIIIB WT donors or the null individual (Figure 3A-B). Apparently, FcγRIIIB-null PMNs were activated slightly more efficiently than FcγRIIIB WT PMNs, although the difference was not statistically significant (Figure 3A-B). Similar data were obtained using unmodified RTX as stimulator instead of RTX-GE (data not shown). Finally, to exclude that the PMNs in the assay were activated indirectly (ie, that the MAbs induced FcγRIIIA+ NK cells or monocytes present in the CLL cell preparation to release cytokines, which, in turn activate PMNs), we also performed activation experiments in Transwells. We could show that direct contact between opsonized CLL B cells and PMNs was necessary for greater activation of the latter by RTX-GE compared with RTX (Figure 3C).

Dose-response curves and contact-dependent activation of PMNs. (A-B) Purified PMNs from FcγRIIIB WT donors and the null individual were stimulated for 4 hours with CLL B cells opsonized with increasing concentrations of RTX-GE. CD11b (A) and CD62L (B) expression on PMNs was analyzed by flow cytometry. (C) FcγRIIIB WT PMNs were activated with CLL B cells opsonized with RTX, RTX-GE, or CTX (CTRL), either together in standard 24-well plates or separated from each other in Transwells. The data are the means and standard deviations of 3 or 4 experiments. *P < .05 vs control (CTRL).
Dose-response curves and contact-dependent activation of PMNs. (A-B) Purified PMNs from FcγRIIIB WT donors and the null individual were stimulated for 4 hours with CLL B cells opsonized with increasing concentrations of RTX-GE. CD11b (A) and CD62L (B) expression on PMNs was analyzed by flow cytometry. (C) FcγRIIIB WT PMNs were activated with CLL B cells opsonized with RTX, RTX-GE, or CTX (CTRL), either together in standard 24-well plates or separated from each other in Transwells. The data are the means and standard deviations of 3 or 4 experiments. *P < .05 vs control (CTRL).
We conclude that enhanced PMN activation by glycoengineered antibodies compared with unmodified antibodies does not require FcγRIIIB.
PMNs from FcγRIIIB WT and null donors express FcγRIIIA protein and messenger RNA
In search of a possible explanation for our results, we noted that anti-CD16 antibodies show a very weak, but not completely negative, staining of PMNs from the FcγRIIIB-null donor (see the 2% positivity in Figure 1B). Therefore, we hypothesized that low levels of FcγRIIIA might be expressed by PMNs in the FcγRIIIB-null donor. We first repeated the flow cytometry analyses using a panel of anti-CD16 antibodies that detect FcγRIIIA and FcγRIIIB. As shown in Figure 4A, although CD16 was highly expressed on PMNs from FcγRIIIB WT donors, all CD16 antibodies used revealed a very low, but detectable, expression of FcγRIII on PMNs from the FcγRIIIB-null donor compared with isotype controls. The positivity was seen as a shift of the curve in all cases, indicating that all cells are very weakly positive. This suggested that PMNs from the FcγRIIIB-null donor may indeed express some FcγRIIIA on their surfaces, albeit at rather low levels.
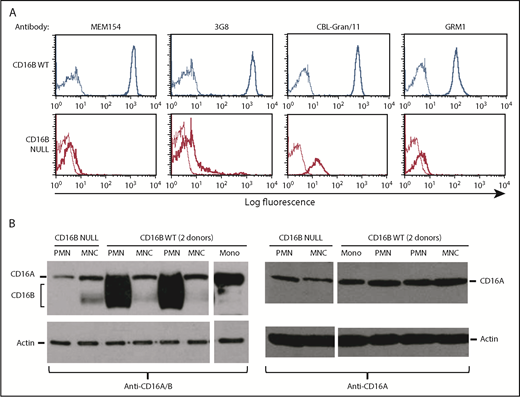
Purified PMNs express FcγRIIIA protein. (A) Purified PMNs from FcγRIIIB WT donors and the FcγRIIIB-null individual were stained with FITC-labeled (clones MEM154 and 3G8; both mIgG1) or unlabeled (clones CBL-Gran/11 and GRM1; both mIgG2a) anti-CD16 antibodies, followed by goat anti-mouse IgG–FITC (thick lines). Appropriate negative controls were used in each case (thin lines). (B) Purified MNCs, PMNs, or monocytes (Mono) isolated from 2 FcγRIIIB WT donors and the null individual were lysed and western blot analysis was performed using antibodies recognizing FcγRIIIA and FcγRIIIB (left panel) or specific for FcγRIIIA only (right panel). Actin antibody was used as control. The indicated bands for FcγRIIIB, FcγRIIIA, and actin were 50-75 kDa, 70 kDa, and 45 kDa, respectively.33 The results are representative of at least 3 experiments.
Purified PMNs express FcγRIIIA protein. (A) Purified PMNs from FcγRIIIB WT donors and the FcγRIIIB-null individual were stained with FITC-labeled (clones MEM154 and 3G8; both mIgG1) or unlabeled (clones CBL-Gran/11 and GRM1; both mIgG2a) anti-CD16 antibodies, followed by goat anti-mouse IgG–FITC (thick lines). Appropriate negative controls were used in each case (thin lines). (B) Purified MNCs, PMNs, or monocytes (Mono) isolated from 2 FcγRIIIB WT donors and the null individual were lysed and western blot analysis was performed using antibodies recognizing FcγRIIIA and FcγRIIIB (left panel) or specific for FcγRIIIA only (right panel). Actin antibody was used as control. The indicated bands for FcγRIIIB, FcγRIIIA, and actin were 50-75 kDa, 70 kDa, and 45 kDa, respectively.33 The results are representative of at least 3 experiments.
We then performed western blots of the FcγRIII proteins. Using an antibody that recognizes FcγRIIIA and FcγRIIIB, we could see that, as expected, neutrophils from 2 FcγRIIIB WT donors, but not the FcγRIIIB-null subject, expressed a strong 50- to 75-kDa signal, corresponding to FcγRIIIB, a highly glycosylated protein32 (Figure 4B, left panel). In contrast, MNCs from all donors expressed a narrower 70-kDa band corresponding to FcγRIIIA.33 This same band was also present in the PMNs from the FcγRIIIB-null donor. Of note is that a 60- to 65-kDa band was observed with anti-CD16A/B antibody in some MNC samples, including those derived from the FcγRIIIB-null donor, but not in PMNs (Figure 4B, left panel). This band may represent a nonspecific band or an alternative isoform of CD16A, but it has not been investigated further. When using an antibody specific for the FcγRIIIA intracellular cytoplasmic tail, we could detect the FcγRIIIA 70-kDa protein in MNCs and monocytes, as expected, as well as in PMNs from all donors (FcγRIIIB WT and null) (Figure 4B, right panel). We conclude that PMNs do express FcγRIIIA, albeit at low levels, and that the FcγRIIIA protein is more difficult to detect by flow cytometry and western blot in the presence of large amounts of FcγRIIIB.
To confirm, at the RNA level, that PMNs express FcγRIIIA, we performed a double-purification step to obtain PMNs with >99.5% purity and with ≤0.1% monocytes and ≤0.3% lymphocytes. We designed several sets of primers to specifically amplify the FCGR3A and FCGR3B transcripts by RT-PCR. The products were also digested with TaqI enzyme to further distinguish between FCGR3A and FCGR3B products. Using this strategy, we could detect the FCGR3B-specific bands in FCGR3B WT PMNs but not in PMNs from the null donor, as expected. In contrast, the FCGR3A bands were amplified in PMNs from all donors, as well as in monocytes or MNCs, as would be expected (Figure 5).

Highly purified PMNs express FCGR3A messenger RNA. PMNs from 6 FcγRIIIB WT donors (WT1-6), as well as the FcγRIIIB-null donor (Null), were purified to >99%. Peripheral blood MNCs or CD14+ purified monocytes (Mono) were used as controls. RNA was extracted, and RT-PCR was performed using FCGR3A- or FCGR3B-specific primers, corresponding to exon 5 (A) or exon 3 to 4 (B) sequences. PCR products were cut with TaqI restriction enzyme to distinguish the 2 genes and run on a 4% agarose gel. The expected products for FCGR3A or FCGR3B are shown on the right.
Highly purified PMNs express FCGR3A messenger RNA. PMNs from 6 FcγRIIIB WT donors (WT1-6), as well as the FcγRIIIB-null donor (Null), were purified to >99%. Peripheral blood MNCs or CD14+ purified monocytes (Mono) were used as controls. RNA was extracted, and RT-PCR was performed using FCGR3A- or FCGR3B-specific primers, corresponding to exon 5 (A) or exon 3 to 4 (B) sequences. PCR products were cut with TaqI restriction enzyme to distinguish the 2 genes and run on a 4% agarose gel. The expected products for FCGR3A or FCGR3B are shown on the right.
Finally, we performed quantitative PCR using probes specific for FCGR3A and FGGRB. The PMNs from WT donor 5 expressed a calculated 12.6-fold less FCGR3A than FCGR3B messenger RNA (data not shown).
We conclude that PMNs express low levels of FCGR3A transcript and protein, which are usually masked by high FCGR3B expression.
FcγRIIIA is sufficient for PMN activation and phagocytosis
The enhanced activation on PMNs by glycoengineered anti-CD20 antibodies shown above suggested that FcγRIIIA is the signaling molecule in this case. To demonstrate this directly, we used anti-CD16 and anti-CD32 antibodies. We observed efficient PMN activation, detected as CD11b induction, by F(ab′)2 or whole anti-CD16 antibodies, but not anti-CD32 antibodies, whether PMNs were derived from FcγRIIIB WT or null donors (Figure 6A). Similarly, we observed upregulation of the activation-specific CD11b neoepitope, detected by CBRM1/5 antibody clone,34 following anti-CD16 stimulation (supplemental Figure 1). This suggests that FcγRIIIA cross-linking is sufficient for PMN activation and that FcγRIIIA is functional, because this is the only FcγRIII expressed in the null donor.
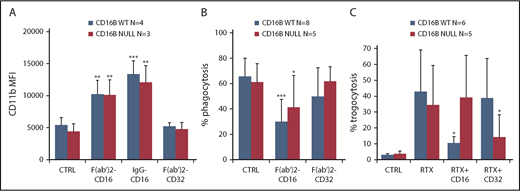
FcγRIIIA is functional on PMNs. PMNs were purified from FcγRIIIB WT donors and the null individual, and their functional activity was measured. (A) Activation was measured by CD11b induction after stimulation with 10 µg/mL anti-CD16 F(ab′)2, whole antibody (MAb), or anti-CD32 F(ab′)2 antibodies. (B) Phagocytosis was measured by coincubation of PMNs for 1 hour with RTX-coated beads in the absence (CTRL) or presence of 10 µg/mL anti-CD16 F(ab′)2 or anti-CD32 F(ab′)2 antibodies. The percentage of PMNs having phagocytosed ≥1 bead was measured. (C) Trogocytosis was measured by coincubation of purified PMNs with PKH67-labeled CLL B cells in the presence of 1 µg/mL RTX and presence or absence of 10 µg/mL blocking anti-CD32 and anti-CD16 IgG antibodies. The percentage of PKH67+ PMNs was then measured after 4 hours by flow cytometry. The results are the means and standard deviations of 5 to 8 independent experiments. *P < .05, **P < .01, ***P < .001 vs the respective controls without antibody (CTRL).
FcγRIIIA is functional on PMNs. PMNs were purified from FcγRIIIB WT donors and the null individual, and their functional activity was measured. (A) Activation was measured by CD11b induction after stimulation with 10 µg/mL anti-CD16 F(ab′)2, whole antibody (MAb), or anti-CD32 F(ab′)2 antibodies. (B) Phagocytosis was measured by coincubation of PMNs for 1 hour with RTX-coated beads in the absence (CTRL) or presence of 10 µg/mL anti-CD16 F(ab′)2 or anti-CD32 F(ab′)2 antibodies. The percentage of PMNs having phagocytosed ≥1 bead was measured. (C) Trogocytosis was measured by coincubation of purified PMNs with PKH67-labeled CLL B cells in the presence of 1 µg/mL RTX and presence or absence of 10 µg/mL blocking anti-CD32 and anti-CD16 IgG antibodies. The percentage of PKH67+ PMNs was then measured after 4 hours by flow cytometry. The results are the means and standard deviations of 5 to 8 independent experiments. *P < .05, **P < .01, ***P < .001 vs the respective controls without antibody (CTRL).
An important function of PMNs is phagocytosis. To measure phagocytosis, we used RTX-coated 5-µm beads, because we had previously observed that RTX-opsonized CLL B cells are too large to be phagocytosed by PMNs.35 We performed phagocytosis assays using purified PMNs isolated from FcγRIIIB WT donors or the null individual. As shown in Figure 6B, PMNs from FcγRIIIB WT or null donors were able to phagocytose the beads to a similar extent; in both cases, the anti-CD16 antibodies, but not the anti-CD32 F(ab′)2 antibodies, were able to inhibit phagocytosis. Similar results were obtained when the phagocytic index, rather than the percentage of phagocytosing cells, was measured (data not shown). In support of the specificity of the assay was the finding that anti-CD16 or anti-CD32 antibodies did not affect the phagocytosis of fibronectin-coated beads used as control (data not shown).
These data indicate that FcγRIIIA is an important mediator of PMN activation and phagocytosis.
FcγRIIIB mediates trogocytosis
We have shown previously that PMNs also mediate trogocytosis of antibody-opsonized CLL target cells.11 Therefore, we analyzed the roles of FcγRIIIA/B and FcγRIIA in this phenomenon. PMNs from FcγRIIIB WT donors and the null individual were able to mediate trogocytosis (Figure 6C). FcγRIIIB-null PMNs mediated somewhat lower trogocytosis than WT PMNs, but this was not statistically significant in these experiments. CD16 antibody significantly inhibited trogocytosis of RTX-opsonized CLL B cells by FcγRIIIB WT PMNs, but not by FcγRIIIB-null PMNs, demonstrating the role of FcγRIIIB in trogocytosis (Figure 6C) (P < .05). In contrast, anti-CD32 was able to partially block FcγRIIIB-null PMN–mediated trogocytosis, but not WT PMN–mediated trogocytosis (P < .05), showing that, in the absence of FcγRIIIB, FcγRIIA can mediate trogocytosis (Figure 6C).
Altogether, these data suggest that FcγRIIIB is a major mediator of trogocytosis by FcγRIIIB WT PMNs, and FcγRIIA performs this function in FcγRIIIB-null PMNs.
Discussion
In this study, we have identified a very rare FcγRIIIB-null donor. Absence of FcγRIIIB in this donor’s PMNs was demonstrated by the lack of FCGR3B DNA and RNA amplification by PCR and the lack of FcγRIIIB protein expression by flow cytometry and western blot. Several previous FcγRIIIB-null donors have been described in the literature who have a deletion of CD16B and CD32C genes due to homologous recombination.27-29,36
The availability of a rare natural knockout for FcγRIIIB allowed us to investigate in more detail the role of this molecule in PMN functions induced by therapeutic anti-CD20 antibodies. We first demonstrated that FcγRIIIB was fully dispensable for PMN activation by opsonized targets, because neutrophils were activated equally well whether they derived from FcγRIIIB WT or null donors. The fact that FcγRIIIB-null PMNs were activated more strongly by glycoengineered anti-CD20 antibodies compared with fully core-fucosylated anti-CD20 antibodies also gave us the hint that FcγRIIIA may be present on PMNs, because defucosylated antibodies have a higher affinity for FcγRIIIA and FcγRIIIB but not for other FcγRs.11,13,14,37,38 Further investigation fully demonstrated the presence of low levels of FcγRIIIA on PMNs from all donors. This was shown by RT-PCR using different primers, RT–quantitative PCR with specific probes, protein detection on FcγRIIIB-null PMNs by flow cytometry, and protein detection by western blot on all PMNs using FcγRIIIA-specific antibody. The very low levels of FcγRIIIA protein expression compared with FcγRIIIB protein expression on PMNs, the high homology between these 2 genes and protein products, and the fact that the wide 50- to 75-kDa FcγRIIIB band masks 70-kDa FcγRIIIA protein in western blots are the probable explanations for the missed identification of FcγRIIIA on PMNs previously. We believe that this is the first report of FcγRIIIA expression on PMNs.
Therefore, FcγRIIIA expression on PMNs may explain their greater activation by glycoengineered anti-CD20 MAbs compared with WT CD20 MAbs, even in absence of FcγRIIIB. Of note is that FcγRIIIA has an eightfold to 10-fold higher affinity for IgG1 than does FcγRIIIB,39 allowing binding and signaling by FcγRIIIA, even in the presence of higher levels of FcγRIIIB. That FcγRIIIA expressed by PMNs is functional was shown directly, because anti-CD16 F(ab′)2 antibody was sufficient to activate PMNs from the FcγRIIIB-null donor. These data may explain the previous observations of PMN activation by anti-CD16 antibodies, which had been ascribed to FcγRIIIB through a not very well–defined mechanism, because this molecule is glycosylphosphatidylinositol linked and completely lacks a signaling module or signaling capacity by itself.23,40-43 Indeed, several groups have suggested cooperation between FcγRIIIB and FcγRIIA or CD18 for signaling.26,44,45 In contrast to those previous studies, we suggest that FcγRIIIA may be the activating CD16 family receptor on PMNs. However, further direct demonstration of this point will require a detailed analysis of the signaling pathways that are triggered by ligand binding to FcγRIIIA and FcγRIIA in CD16B WT and null donors. These studies are beyond the scope of this article.
We were also interested in establishing the role of FcγRIIIA and FcγRIIIB in some PMN functions. With regard to phagocytosis, we observed an equivalent capacity of FcγRIIIB WT and null PMNs to phagocytose RTX-coated beads, suggesting that FcγRIIIB is dispensable for phagocytosis. Indeed, further blocking experiments with anti-CD16 F(ab′)2 antibodies indicated that FcγRIIIA does indeed play a role in the phagocytosis of antibody-opsonized beads by PMNs. In contrast, F(ab′)2 anti-CD32 did not significantly affect phagocytosis in our assay. These data are in agreement with a previous report showing that FcγRIIIB is fully dispensable for phagocytosis of IgG-opsonized bacteria or reactive oxygen species activation using a similar FcγRIIIB-null donor.36 Also, PMNs from patients with paroxysmal nocturnal hemoglobinuria almost completely lack FcγRIIIB, and the study of these cells has also indicated that FcγRIIIB is dispensable for some PMN responses, although the interpretation of results is more difficult in this case due to residual FcγRIIIB expression.44,46,47 In particular, data from 1 group using anti-EGFR antibodies optimized for FcγRIII binding, as well as PMNs from paroxysmal nocturnal hemoglobinuria patients, suggested that FcγRIIA is the major mediator of ADCC by PMNs and that FcγRIIIB may act as a decoy receptor in this case.10,48 Similarly, the role of FcγRIIIB in PMNs has been studied after its removal using phosphodylinositol–phospholipase C treatment. In this case, FcγRIIIB appeared to be required for respiratory burst but not for phagocytosis and killing of IgG-opsonized bacteria.49
Another phenomenon mediated by PMNs is trogocytosis of antibody-opsonized cells.11,50 Anti-CD16 antibodies only blocked trogocytosis of FcγRIIIB WT PMNs, suggesting an important role for FcγRIIIB in this phenomenon. FcγRIIIB is normally a very abundant protein on the PMN surface and is likely therefore to mediate close and strong interaction between effector and target cells. This strong cell-cell binding may be necessary for efficient trogocytosis and may explain the role of FcγRIIIB in this phenomenon.6,40,50-52 In contrast, FcγRIIA was primarily responsible for trogocytosis by FcγRIIIB-null PMNs, presumably due to the fact that it is the most abundant FcγR on these cells. This observation again suggests that the strength of binding between 2 cells may be the main determinant for trogocytosis; however, this will need to be confirmed by specific studies that also investigate the role of intracellular signaling in this phenomenon. Interestingly, FcγRIIIB on neutrophils dampens the ADCC activity mediated by EGFR antibodies, suggesting a negative effect of FcγRIIIB in some circumstances.10
Thus, our data and those from the available literature suggest that the major role of FcγRIIIB is to bind IC and mediate perhaps only some functions, such as trogocytosis and endocytosis. Indeed, the fact that FcγRIIIB-null donors are healthy and do not have increased infections supports a marginal role for FcγRIIIB in the control of infections.28 Clearly, further studies will be required to more fully dissect the role of FcγRIIIB and other FcγRs in all major PMN functions in the presence of a panel of different antibodies, not only anti-CD20 MAbs. Most importantly, early signaling events downstream of FcγRIIIA and FcγRIIA also need to be fully dissected. The identification of other FcγRIIIB-null donors appears to be necessary to achieve such a task. Similarly, the study of possible expression of low levels of FcγRIIIA in PMNs of transgenic mice or of cynomolgus monkeys, which lack CD16B, is warranted to generate tools to further dissect the role of this family of receptors in PMN functions.53,54
Figure 7 summarizes the findings reported in this article: that PMNs express low levels of FcγRIIIA, which nonetheless may be a signaling molecule for neutrophil activation and phagocytosis of IgG-opsonized targets by these cells. In contrast, FcγRIIIB is very abundant on WT PMNs and mediates efficient binding to IgG-opsonized targets and subsequent trogocytosis. Interestingly, polymorphisms of CD16B are weakly associated with some diseases, such as glomerulonephritis and systemic lupus erythematosus.55-58 In agreement with previous data, we suggest that this association may be related to the function of FcγRIIIB in IC clearance following binding and perhaps endocytosis and/or receptor shedding.41,58
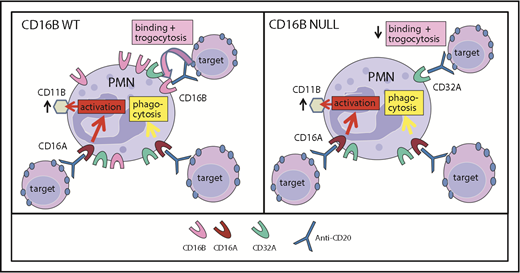
Model of FcγRIIIA and FcγRIIIB function in PMNs. Model of CD16A and CD16B activities in PMN function in FcγRIIIB WT donors (A) and the FcγRIIIB-null individual (B). FcγRIIIA plays a major role in PMN activation (measured as CD11b induction) and phagocytosis. FcγRIIIB is very abundant on FcγRIIIB WT PMNs and has a role in binding ICs favoring trogocytosis. In the absence of FcγRIIIB, trogocytosis takes place primarily via FcγRIIA.
Model of FcγRIIIA and FcγRIIIB function in PMNs. Model of CD16A and CD16B activities in PMN function in FcγRIIIB WT donors (A) and the FcγRIIIB-null individual (B). FcγRIIIA plays a major role in PMN activation (measured as CD11b induction) and phagocytosis. FcγRIIIB is very abundant on FcγRIIIB WT PMNs and has a role in binding ICs favoring trogocytosis. In the absence of FcγRIIIB, trogocytosis takes place primarily via FcγRIIA.
We have not identified a clear role for FcγRIIA in WT PMNs, using blocking antibodies. FcγRIIA has been implicated by other investigators in anti-EGFR–induced ADCC.10 However, anti-CD20 MAbs are unable to mediate this activity,6 so ADCC was not investigated in this study. Nonetheless, FcγRIIA bears ITAM motifs and is a signaling molecule; therefore, it is likely to cooperate with FcγRIIIA in some PMN functions.40,59
Interestingly, mice have a different set of FcγRs, and mouse neutrophils are similar to human PMNs in that they express 2 activating FcγRs (mouse FcγRIII and FcγRIV). In contrast, mice do not have the FCGR3B gene, but their neutrophils express inhibitory FcγRIIB.16
The findings reported here have wide clinical implications in the therapeutic antibody field and in autoimmunity. For example, the presence of functional activating FcγRIIIA on PMNs raises the question as to whether the FcγRIIIA polymorphisms may also affect PMN function in vitro or in vivo in response to therapeutic IgG1 antibody treatment, especially in light of the recent role suggested for neutrophils in this context by different groups.2,3,5 Our findings also open new avenues for dissecting the possible role of FcγRIIIA polymorphisms in antibody-dependent autoimmune diseases or anaphylaxis, in which FcγRIII and/or PMNs have been implicated, such as bullous pemphigoid, rheumatoid arthritis.60-65 Finally, the recent finding that some endogenous pathogenic IgG1 antibodies may be hypofucosylated66 opens the question as to whether FcγRIIIA on neutrophils plays a role in their pathogenic effect.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Christian Klein for the kind gift of RTX-GE. The authors are grateful to all donors who repeatedly provided blood for this study.
This work was funded in part by the Associazione Italiana Ricerca contro il Cancro (AIRC Investigator grant IG 19036 [J.G.] and AIRC 5x1000 ISM) and the Associazione Italiana contro le leucemie-linfomi e mieloma-sezione Paolo Belli, Bergamo.
Authorship
Contribution: R.V. and G.M. performed activation, trogocytosis, and phagocytosis experiments and western blots and analyzed the data; O.S. and D.G. performed genomic DNA PCR; J.G. supervised experiments, performed RT-PCR, and wrote the manuscript; and M.I. provided funding and critically revised the manuscript.
Conflict-of-interest disclosure: J.G. has received research funding and consultancy fees from Roche Glycart. The remaining authors declare no competing financial interests.
Correspondence: Josée Golay, Center of Cellular Therapy “G. Lanzani” and Division of Hematology, Azienda Socio Sanitaria Territoriale Papa Giovanni XXIII, Via Garibaldi 11-13, 24128 Bergamo, Italy; e-mail: jgolay@asst-pg23.it.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal