

Abstract
The RAS-RAF-MEK-ERK (MAPK) pathway is deregulated in many cancers including acute myeloid leukemia (AML), where it facilitates cancer progression, chemo-resistance and cancer relapse. Therefore, the inhibition of this pathway has been explored for the treatment of AML, especially in the ~10% of AML cases where the leukemic cells carry activating mutations of the NRAS or KRAS oncogenes. However, MEK inhibitors, the best explored class of MAPK inhibitors, have not proven effective in the clinical setting. A novel clinically investigated drug class in this context are the paradox-breaker pan-RAF inhibitors that prevent the reactivation of the MAPK-pathway and have exhibited single agent efficacy in preclinical testing for solid cancers. Therefore, it is relevant to explore the utility of this class of drugs for AML treatment.
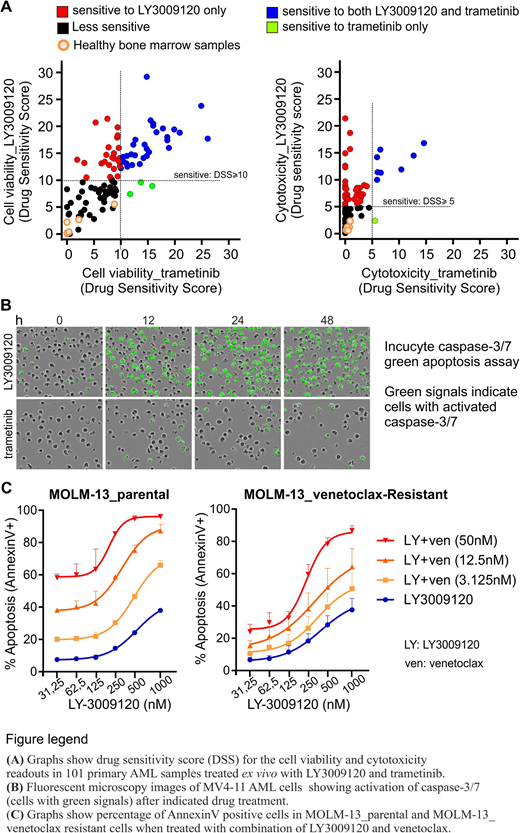
We found that the paradox breaker pan-RAF inhibitors LY3009120 (currently in clinical testing), AZ-628 and TAK-632 as well as other MAPK inhibitors effectively downregulated MAPK signaling activity (ERK phosphorylation) in AML cells. However, when comparing cell viability (CellTiter Glo) and cell death (CellTox Green) responses in 101 AML patient sample cohort, only the pan-RAF inhibitors induced cytotoxicity while the other MAPK-pathway inhibitors generally induced a temporary cytostatic response. Specifically, LY3009120 induced cell death in 42 samples out of 101 (42 %) whereas only 9/101 (9 %) underwent cell death in response to trametinib (MEK-inhibitor) (Fig. panel A). Importantly, LY3009120 did not induce cell death in mononuclear bone marrow cells obtained from healthy individuals (5 samples) or in non-hematopoietic cell lines (Fig. panel A).
To get insight into the mechanism regulating cell death in response to pan-RAF inhibition we first characterized that cells underwent rapid caspase-3/7 mediated apoptotic cell death in response to pan-RAF inhibitors (Fig. panel B). Therefore, we analyzed the proteins involved in apoptosis pathways and found that MCL-1 protein, but not mRNA levels, were downregulated upon treatment with pan-RAF inhibitors but not with MEK-inhibitors. This suggests that pan-RAF inhibition negatively regulated the translation of MCL-1, thus uncovering a new mode of MCL-1 regulation by RAF kinases. Importantly, pan-RAF inhibitors downregulated MCL-1 levels independently of caspase activity, indicating that MCL-1 downregulation precedes the apoptotic response.
We explored the link between somatic mutation status and response to LY3009120 in our AML patient sample cohort. KRAS, KIT, FLT3-TKD, BCOR, JAK2 and PTPN11 mutations were found to correlate positively with pan-RAF inhibitor response, while SRSF2, RUNX1, GATA2 and STAG2 mutations negatively correlated with response and appeared to override positively correlating mutations.
As pan-RAF inhibition downregulates MCL-1, an important survival protein whose increased expression is known to cause resistance to BCL-2 inhibitors, we hypothesized that inhibition of RAF kinases can overcome the resistance to BCL-2 inhibitors. In order to test the hypothesis, we generated venetoclax (BCL-2 inhibitor) resistant MOLM-13 cells and treated them with single and combined doses of venetoclax and LY3009120. As expected, combined inhibition of BCL-2 and RAF kinases significantly increased cytotoxicity as compared to either of the drugs alone (Fig. panel C). Conversely, to see if BCL-2 inhibition can overcome resistance to pan-RAF inhibitors, we treated pan-RAF inhibitor resistant HL-60 and THP-1 cells with single and combined doses of venetoclax and LY3009120. Interestingly, the drug combination exhibited synergistic cytotoxicity in HL-60 cells (where LY3009120 alone downregulates MCL-1) but not in THP-1 cells (where LY3009120 alone does not downregulate MCL-1). This indicates that MCL-1 downregulation upon pan-RAF inhibitor treatment is critical for the observed combination cytotoxicity.
In summary, our results show that paradox breaker pan-RAF inhibitors are effective as single agents to induce cell death in AML blasts, likely through a combined MAPK pathway and MCL-1 dependent mechanism. Moreover, combined inhibition of RAF kinases and BCL-2 can overcome resistance of AML cells to either drug alone and lead to superior anti-leukemic activity.
Heckman:Celgene: Research Funding; Novartis: Research Funding; Orion Pharma: Research Funding. Porkka:Novartis: Honoraria, Research Funding; Celgene: Honoraria, Research Funding. Wennerberg:Novartis: Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal