

Abstract
The ability to upregulate and downregulate surface-exposed proteins and receptors is a powerful process that allows a cell to instantly respond to its microenvironment. In particular, mobile cells in the bloodstream must rapidly react to conditions where infection or inflammation are detected, and become proadhesive, phagocytic, and/or procoagulant. Platelets are one such blood cell that must rapidly acquire and manage proadhesive and procoagulant properties in order to execute their primary function in hemostasis. The regulation of platelet membrane properties is achieved via several mechanisms, one of which involves the controlled metalloproteolytic release of adhesion receptors and other proteins from the platelet surface. Proteolysis effectively lowers receptor density and reduces the reactivity of platelets, and is a mechanism to control robust platelet activation. Recent research has also established clear links between levels of platelet receptors and platelet lifespan. In this review, we will discuss the current knowledge of metalloproteolytic receptor regulation in the vasculature with emphasis on the platelet receptor system to highlight how receptor density can influence both platelet function and platelet survival.
Platelets are involved in a range of vascular responses
Platelets are anucleate cellular elements that circulate in vast numbers through the mammalian vasculature. As part of their premier role in hemostasis, platelets must be able to rapidly move from a resting, noninteractive state, to an adhesive and procoagulant entity. This enables circulating platelets to engage with the blood vessel endothelium, the extracellular matrix, and blood leukocytes directly at the site of injury or infection.1-4
Further to the hemostatic function of platelets, they also make critical contributions to innate immune and inflammatory responses,5-9 lymph and blood vessel development,10,11 tumorigenesis,12 and tumor metastasis.13,14 Platelets help mount the host defense against bacterial invasion,4,5,15-17 through being the first responders that detect pathogens, and new studies reveal that platelets may bind directly to infected erythrocytes and kill intraerythrocytic parasites of malarial Plasmodium species,18 indicative of platelets’ expanding role in innate immune responses. Platelet receptor ectodomains are important molecular players helping to orchestrate many, if not all, of these functions.
Platelet receptors initiate and control platelet function
Platelet receptors are integral to all of these vascular interactions. The transition of circulating quiescent platelets to an active, adhesive state when exposed to extracellular matrix proteins is orchestrated by a range of receptors present on the platelet membrane.19,20 The 2 key adhesion receptors are glycoprotein VI (GPVI), binding collagen, fibrin and fibrinogen, and GPIb-IX-V, which binds von Willebrand factor (VWF) as well as a number of other key ligands. Ligand/receptor engagement triggers activation of signaling pathways that causes changes in calcium flux, the release of soluble agonists, degranulation, and a significant increase in fibrinogen binding via upregulation of αIIbβ3. GPVI is closely associated with GPIbα of the GPIb-IX-V complex,21 and these 2 receptors operate in concert to mediate platelet activation and adhesion events under a variety of blood rheological conditions found in the vasculature.22 An additional consequence of ligand engagement of these receptors is the activation of platelet metalloproteinases. Both of these platelet-specific receptors are exquisitely sensitive to metalloproteolytic regulation, a process where metalloproteinases cleave the receptor, releasing an ectodomain fragment into the plasma.23
Platelet adhesion receptors are metalloproteolytically shed
Platelets are heavily engaged in ectodomain shedding, with some 69 different membrane proteins released in response to treatment with protein kinase C activator phorbol 12-myristate 13-acetate (PMA),24 a generic activator of metalloproteinases. In particular, the metalloproteolytic release of GPIbα,25,26 GPV,27,28 and GPVI29,30 (Figure 1) has been studied extensively by a number of laboratories.31-34 With regard to these receptors, this proteolytic process is mediated by members of the ADAM family and several ADAM family members are detected on platelet membranes. Platelet receptor shedding is reminiscent of the mechanism regulating leukocyte levels of L-selectin,35 an ADAM17-mediated process that enables the detachment of adherent leukocytes from the endothelium and migration to interstitial spaces. Similar to L-selectin shedding, the platelet receptors engage ligands triggering platelet activation, which results in detachment of calmodulin from conserved juxtamembrane-binding motifs within the cytoplasmic tails of the GPIb-IX-V complex (GPIbβ and GPV) and GPVI. Treatment of platelets with calmodulin inhibitors directly activates ADAM-mediated shedding of GPV and GPVI. In platelets, this autolytic process is of great relevance because it represents a mechanism by which the platelet can rapidly modulate surface reactivity, receptor density, and the degree to which platelets engage with hemostatic and inflammation-related ligands. Table 1 provides some details of mechanisms by which proteolysis of platelet receptors have been reported in the literature. Not all receptors have been evaluated with all treatments. By far, the platelet adhesion receptors have been the most widely studied. In the following sections, we will discuss some of the more prolific metalloproteolytically shed proteins from platelets and consider the ramifications of loss of membrane protein ectodomains on platelet function.
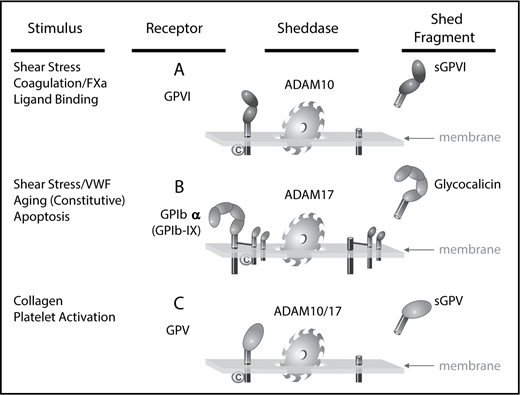
Receptor shedding from human platelets. ADAM10 and ADAM17 are responsible for the shedding of platelet-surface receptor ectodomains of GPIbα, GPV, and GPVI. The physiological triggers of ADAM activity dictate the rate and extent of loss of specific receptors. (A) GPVI is stable on resting platelets but rapidly cleaved by ADAM10 on exposure to active factor X (FXa) or fibrin, as well as by brief exposure to elevated fluid shear stress, and collagen binding. (B) The release of glycocalicin from the GPIb-IX complex occurs constitutively and in response to apoptosis. Shedding is mediated by ADAM17 with implications for platelet lifespan and TPO production. (C) GPV is susceptible to shedding by both ADAM10 and ADAM17 under conditions of collagen binding and platelet activation. ©, calmodulin.
Receptor shedding from human platelets. ADAM10 and ADAM17 are responsible for the shedding of platelet-surface receptor ectodomains of GPIbα, GPV, and GPVI. The physiological triggers of ADAM activity dictate the rate and extent of loss of specific receptors. (A) GPVI is stable on resting platelets but rapidly cleaved by ADAM10 on exposure to active factor X (FXa) or fibrin, as well as by brief exposure to elevated fluid shear stress, and collagen binding. (B) The release of glycocalicin from the GPIb-IX complex occurs constitutively and in response to apoptosis. Shedding is mediated by ADAM17 with implications for platelet lifespan and TPO production. (C) GPV is susceptible to shedding by both ADAM10 and ADAM17 under conditions of collagen binding and platelet activation. ©, calmodulin.
Agonists or treatments reported to metalloproteolytically release platelet membrane receptors
| Physiological agonist | Experimental treatments | |
|---|---|---|
| GPIbα | Shear stress/VWF | Calmodulin inhibition |
| Oxidative stress | N-ethylmaleimide | |
| Mitochondria-aging reagents | ||
| BH3 mimetics | ||
| PMA | ||
| GPV | Thrombin | PMA |
| Calmodulin inhibition | ||
| Oxidative stress | N-ethylmaleimide | |
| Convulxin | ||
| GPVI | Collagen | Calmodulin inhibition |
| Fibrin | N-ethylmaleimide | |
| Shear stress | Mitochondrial-aging reagents | |
| Antiplatelet autoantibodies | BH3 mimetics | |
| Factor Xa | Convulxin | |
| Sema4D | Collagen | PMA |
| PAR-1 agonist | ||
| PKA activation | ||
| Calmodulin inhibition | ||
| CD40L | Collagen | PAR-1 agonist |
| Thrombin | PAR-4 agonist |
| Physiological agonist | Experimental treatments | |
|---|---|---|
| GPIbα | Shear stress/VWF | Calmodulin inhibition |
| Oxidative stress | N-ethylmaleimide | |
| Mitochondria-aging reagents | ||
| BH3 mimetics | ||
| PMA | ||
| GPV | Thrombin | PMA |
| Calmodulin inhibition | ||
| Oxidative stress | N-ethylmaleimide | |
| Convulxin | ||
| GPVI | Collagen | Calmodulin inhibition |
| Fibrin | N-ethylmaleimide | |
| Shear stress | Mitochondrial-aging reagents | |
| Antiplatelet autoantibodies | BH3 mimetics | |
| Factor Xa | Convulxin | |
| Sema4D | Collagen | PMA |
| PAR-1 agonist | ||
| PKA activation | ||
| Calmodulin inhibition | ||
| CD40L | Collagen | PAR-1 agonist |
| Thrombin | PAR-4 agonist |
BH3, Bcl-2 homology domain 3; PAR, protease-activated receptor; PKA, protein kinase A.
GPIbα
GPIbα is the ligand-binding portion of the GPIb-IX-V complex. It is a member of the leucine-rich repeat family and is heavily glycosylated. GPIbα is abundant on the platelet surface with copy numbers estimated by mass spectrometry to be ∼20 000 per platelet.36 The ectodomain of GPIbα (termed glycocalicin) is long known to be released from platelets37 and relatively high (1-3 μg/mL) levels of glycocalicin are found in plasma from healthy donors consistent with at least a portion of GPIbα being constitutively shed from platelets in vivo. Examination of human washed platelets that were collected and maintained in buffers containing metalloproteinase inhibitors, by western blot using an antibody against the cytoplasmic tail of GPIbα detecting intact and cleaved GPIbα, revealed at least 3 forms of this subunit (termed intact, N-terminally clipped, and a remnant C-terminal fragment) on untreated, circulating platelets.26 The process leading to formation of the N-terminally clipped form of GPIbα has not been specifically described, however, the anionic region containing 3 sulfated tyrosine residues has been shown to be susceptible to cleavage by cathepsin G and other proteases.38,39
The majority of GPIbα shed from human and mouse platelets, forming the glycocalicin fragment, was shown to be mediated by ADAM1725,26 (also known as tumor necrosis factor-α–converting enzyme) and the cleavage site within human GPIbα was identified.26 Interestingly, this ADAM17-cleaved sequence lies within a mechanosensory region40 important for fluid shear resistance of VWF-platelet interactions. Thus, metalloproteolysis of GPIbα could be regulated by VWF engagement of GPIbα as well as the biorheological environment. GPIbα is also released from platelets in transfusion bags and the loss of GPIbα may be directly linked to the platelet storage lesion.41-44 Constitutive proteolysis of GPIbα is likely to be a signature event of platelet aging as treatment with artificial aging agents induces GPIbα release,45,46 and GPIbα proteolysis from stored platelets contributes to the accelerated removal of transfused platelets.42,47 Kinase inhibitors47 as well as antibodies that specifically block metalloproteolytic release of GPIbα42 prolong the lifespan of transfused platelets in mouse transfusion models. Although it remains to be conclusively shown that loss of GPIbα ectodomain determines the lifespan of a circulating platelet, this idea is consistent with the notion that the degree of glycosylation of GPIbα is involved in the clearance of platelets within the liver.48 Metalloproteolytic release of glycocalicin would achieve an overall reduction in surface density of GPIbα and the amount of platelet surface glycosylation. Most treatments (Table 1) resulted in the generation of the glycocalicin fragment of ∼130 kDa and increasing amounts of a cytoplasmic tail remnant fragment. This tail fragment is likely to remain associated with the platelet cytoskeleton, thus stabilizing the fragment, protecting it from degradation and enabling detection by western blot. This stability enabled detection of shedding of GPIbα from infiltrating activated platelets in lysates of brain tissue taken from mice that had undergone focal neurotrauma.49
GPV
Although the GPIbα and GPIbβ subunits are stabilized by disulfide bonds and links with the platelet cytoskeleton, GPV is only weakly associated with GPIbα most likely through interactions between transmembrane helices.50 GPV binds collagen and may also enhance thrombin binding to GPIbα. GPV knockout mice had a decreased tendency to form occluding thrombi in an intravital arterial thrombosis model and displayed abnormal platelet interaction with the subendothelium, but no severe bleeding phenotype.51 In studies in vitro, GPV-deficient platelets exhibited defective adhesion to a collagen type I–coated surface under flow or static conditions. GPV can be cleaved by both ADAM10 and ADAM17 at a juxtamembrane region as well as by thrombin at a separate ectodomain site.26,27,52 Analysis of the soluble ectodomain fragment of GPV (sGPV) in plasma revealed significantly increased levels during acute phases of unstable angina pectoris, indicating that sGPV may be a useful marker of platelet activation in those patients.53
GPVI
GPVI helps accelerate thrombin generation at the platelet surface,54,55 and as well as binding collagen and laminin, has the ability to bind polymerized fibrin55,56 and fibrinogen.57,58 Apart from initiation of important signaling processes leading to the upregulation of αIIbβ3 function, interaction with some/all of these ligands enhances thrombus stability. On untreated human platelets, GPVI has ∼9600 copies36 and levels are stable with minimal evidence of constitutive cleavage.26 However, under conditions of exposure to GPVI ligands,30,59 pathophysiological shear stress,60 platelet-activating antibodies,61,62 or activation of coagulation,63 GPVI is cleaved primarily by ADAM10, to generate a soluble ∼50- to 55-kDa ectodomain fragment (termed sGPVI) and a ∼10-kDa cytoplasmic domain fragment (Figure 1). The main metalloproteinase responsible for release of GPVI from murine platelets is less clear with roles for ADAM10 and ADAM17 and possibly other metalloproteinases identified.64 Congenital and autoimmune-based acquired deficiency of GPVI in humans appears to be rare, with only isolated cases reported in the literature,65 however, platelets from these patients do not respond to collagen and these individuals suffer a mild to moderate bleeding diathesis.61,62 Abnormal platelet GPVI expression/collagen responsiveness may also be associated with myeloid/lymphoid bone marrow defects.66,67
Mice deficient in GPVI or the associated FcRγ chain, which is obligatory for GPVI expression, have normal platelet size and number and minimal disruption to normal hemostasis.68 However, depletion or deficiency of GPVI conferred protection in thrombosis models, particularly in models that used exposure of collagen (eg, ferric chloride injury)69 to trigger thrombosis. In systems where thrombus stability was evaluated, loss of GPVI resulted in inability to maintain stable occlusion.56,70 It seems likely that the reduced thrombus stability in mice bearing GPVI-deficient platelets results from a combination of reduced collagen and fibrin binding, and reduced capacity to produce thrombin. Antibody-mediated shedding of GPVI in mice injected with Klebsiella pneumoniae to mimic sepsis, compromised local host defenses and accelerated bacterial infection to the lung and bronchoalveolar,71 suggesting that removal of GPVI impacted on local immunity. It is likely that the GPVI contribution to immunity varies according to the site and nature of the inflammatory stimulus, as has been shown for GPVI and other platelet receptor contributions to inflammatory hemostasis.72
GPVI signaling is sensitive to receptor density,73 which can be modulated by ligand-induced clustering74 and dimerization.75-77 The surface density of GPVI, for example, may be a consequence of both expression levels (regulated by individual genotype and/or disease factors) and the extent of extracellular shedding (regulated by a number of potential factors, as discussed in ”Consequences of platelet receptor shedding”), with expression levels also potentially influencing corresponding levels of sGPVI in plasma (Figure 2). The relationship between surface GPVI density and plasma sGPVI levels in healthy or disease states is complex,78 and elevated plasma sGPVI levels could reflect high surface expression (increased thrombotic risk) or high shedding/low surface levels (increased bleeding risk). Whether increased shedding might represent a compensatory mechanism under prothrombotic conditions, or how low surface GPVI/high plasma sGPVI is related to increased bleeding risk requires further evaluation (Figure 2). Further to this, GPVI is important for an appropriate host response and to minimize inflammation-related bleeding in a range of inflammation models.79 Taken together, available data indicate that intact GPVI is important for a number of platelet roles and loss of the ligand-binding portion of GPVI significantly impacts platelet activation and function.
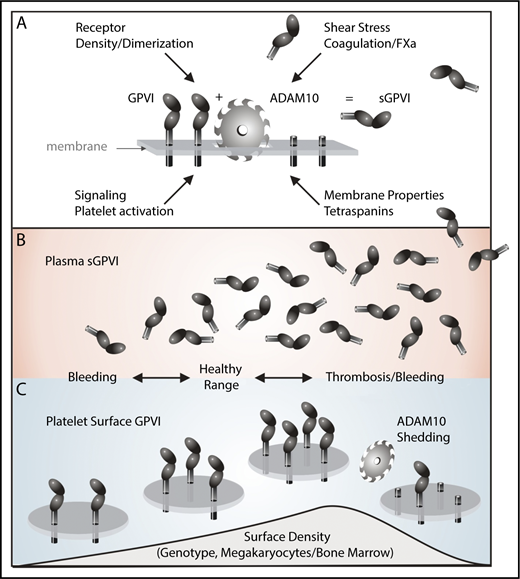
ADAM10-mediated shedding of human platelet GPVI. (A) GPVI on the platelet surface is a 62-kDa transmembrane glycoprotein with 2 extracellular immunoglobulin domains, a short mucin-like domain, a transmembrane domain, and a cytoplasmic tail. Following activation of the metalloproteinase ADAM10 by exposure to elevated shear stress, coagulation, or GPVI ligands, an ∼55-kDa ectodomain of sGPVI is irreversibly shed, leaving a remnant membrane-associated fragment. Membrane properties/tetraspanin expression may also regulate ADAM10 activity toward GPVI. Platelet surface density of GPVI is crucial for signaling/adhesive function to collagen and thrombus stability via GPVI/fibrin. (B-C) Although surface density of GPVI may depend on multiple causes including genotype and/or bone marrow defects, and expression levels may in turn influence plasma levels of sGPVI, higher surface density may be related to increased thrombotic risk, whereas abnormally low levels of GPVI due to deficiency or disease, enhanced ADAM10-mediated shedding together with elevated plasma sGPVI may be associated with an increased risk of bleeding.
ADAM10-mediated shedding of human platelet GPVI. (A) GPVI on the platelet surface is a 62-kDa transmembrane glycoprotein with 2 extracellular immunoglobulin domains, a short mucin-like domain, a transmembrane domain, and a cytoplasmic tail. Following activation of the metalloproteinase ADAM10 by exposure to elevated shear stress, coagulation, or GPVI ligands, an ∼55-kDa ectodomain of sGPVI is irreversibly shed, leaving a remnant membrane-associated fragment. Membrane properties/tetraspanin expression may also regulate ADAM10 activity toward GPVI. Platelet surface density of GPVI is crucial for signaling/adhesive function to collagen and thrombus stability via GPVI/fibrin. (B-C) Although surface density of GPVI may depend on multiple causes including genotype and/or bone marrow defects, and expression levels may in turn influence plasma levels of sGPVI, higher surface density may be related to increased thrombotic risk, whereas abnormally low levels of GPVI due to deficiency or disease, enhanced ADAM10-mediated shedding together with elevated plasma sGPVI may be associated with an increased risk of bleeding.
Amyloid precursor protein
Platelets contain a significant amount of amyloid precursor protein (APP) on the membrane and stored in granules,80 and are the major source of APP found in plasma.81 The precise role of APP in platelet function has not been ascertained, but APP and/or APP-derived peptides, soluble APPα (sAPPα), or Aβ82 may contribute to platelet-mediated adhesion at sites of cerebral vascular bleeds, and platelets were hyperadhesive in a murine model of Alzheimer disease.83 When added to platelets in vitro, the Aβ isoform of APP stimulates an adenosine 5′-diphosphate–dependent release of Ca2+, activation of platelet signaling pathways; in appropriate experimental conditions, Aβ triggers thrombus formation.84,85 Furthermore, platelets contain all of the secretases required to proteolyze APP to soluble forms, and, notably, sAPPα is produced by the action of ADAM10 or ADAM17, both of which exhibit α-secretase activity. Platelet APP proteolysis is also regulated by calmodulin dissociation.86 In a mass spectrometry study of membrane proteins proteolytically shed from platelets, the amyloid Aβ fragment of APP was detected at high levels,24 and platelets from patients with Alzheimer disease retain significant amounts of APP.87 Although the role and processing of platelet APP remain to be fully elucidated, it is enticing to speculate that activated platelets release metalloproteolysed forms of APP that can accumulate and contribute to amyloid plaques. In this scenario, enhancing platelet ADAM10/17 activity to drive formation of nonamyloidogenic sAPPα and minimize the potential of the β-secretase to generate Aβ peptide is of great research interest.88 Also, in this regard, Alzheimer disease patient plasma contains significantly lower levels of plasma sGPVI,89 consistent with lower platelet ADAM10.88,90
Semaphorin 4D
Semaphorin 4D (Sema4D) is a type 1 single transmembrane protein expressed as a disulfide-linked homodimer on platelets as well as B and T lymphocytes, with roles in B-cell development. From a platelet perspective, Sema4D has been shown to amplify thrombus formation by binding to receptors CD72 and Plexin-B1 on adjacent platelets, triggering contact-dependent signaling when activated platelets come into close proximity91 such as in a thrombus. Upon activation, calmodulin binds and detaches from a site within the Sema4D cytoplasmic tail and significant amounts of the extracellular domain of Sema4D are shed by ADAM17 from the surface of activated platelets as a soluble 120-kDa fragment.92 Of interest, this soluble fragment retains angiogenic activity toward endothelial cells and is a clear example of platelet ADAM activity impacting platelet and vascular biology. Both platelet-associated and soluble Sema4D engage CD72 and Plexin-B1 on lymphocytes, platelets, and endothelial cells, and can trigger proangiogenic and wound-healing responses. Mice deficient in Sema4D exhibited delayed arterial occlusion after vascular injury in vivo, and their platelets showed impaired response to collagen,93 most likely due to disruptions in phosphorylation events downstream of Syk in the ITAM-signaling pathway.91
CD40L
CD40L is a type I transmembrane protein that belongs to the tumor necrosis factor superfamily. It is present on platelet membranes and in granules, and in a number of leukocyte subpopulations where it has a significant role in both innate and adaptive immunity. The majority of soluble CD40L (sCD40L) found in plasma is released not only from activated platelets by metalloproteolytic shedding most likely via the action of matrix metalloproteinases 2 (MMP-2)94 and 9 (MMP-9),95 but also possibly by ADAM17.96 Once released, sCD40L is a platelet agonist and binds to αIIbβ3, triggering outside-in signaling and enhancing thrombus stability.97,98 sCD40L also has a causative role in transfusion-related injury.99
The metalloproteinases regulating platelet receptors
ADAMs
Vascular proteolytic release is generally executed by metalloproteinases, the largest class of catalytic enzymes with the greatest abundance and diversity in the human genome.100 Table 2 highlights the metalloproteinases and inhibitors found within platelets and megakaryocytes. Within that class, the ADAM family of ∼40 mostly membrane-anchored enzymes are responsible for the juxtamembranous release of most cell-surface protein ectodomains.101 A number of laboratories have demonstrated roles for platelet ADAMs to rapidly downregulate levels of platelet adhesion receptors.23,102,103 ADAM10 and ADAM17 chiefly engage in these platelet processes and also cleave many substrates with diverse function within the vasculature,104,105 despite some differences between the cleavage site specificities of ADAM10 and ADAM17 particularly at the P1′ position.106 Noncatalytic interactions of the substrate with the cysteine-rich domain of the ADAM position the substrate for membrane-proximal cleavage.107,108
Platelet-associated metalloproteinases and inhibitors
| MKs | Platelet granules | Platelet membrane | Interacts with | Effect on platelet function | |
|---|---|---|---|---|---|
| ADAM10 | Yes | ND | Yes | Tetraspanins | Release of GPVI, APP |
| ADAM17 | Yes | ND | Yes | Release of GPIbα, GPV, Sema4D, APP, sCD40L? | |
| MMP-1 | Yes | Yes | Yes | αIIbβ3, PAR-1 | Accelerates thrombus formation |
| α2β1 | Primes platelets, cleaves PAR-1, activates platelet signaling | ||||
| MMP-2 | Yes | Yes | Yes | GPIb-IX-V | Increase thrombus formation, cleaves talin |
| αIIbβ3 | Cleaves PAR-1; release of sCD40L? | ||||
| MMP-3 | Yes | Unknown | |||
| MMP-9 | Yes | Yes/No | Yes | Decreased activation; reduced Ca2+ mobilization | |
| Increased thrombus area in MMP-9−\− mice | |||||
| MMP-13 | GPVI, αIIbβ3 | Impaired platelet aggregation to low dose collagen, CRP | |||
| Reduced thrombus formation | |||||
| MMP-14 | Yes | Yes | MMP-2 | Inhibits thrombus growth and stability | |
| TIMP-2 | |||||
| TIMP-1 | Yes | Yes | Inhibits platelet ADAMs? | ||
| TIMP-2 | Yes | Yes | MMP-14 | Synthesized de novo upon activation; inhibits all MMPs | |
| TIMP-3 | Yes | Inhibits platelet ADAMs? | |||
| TIMP-4 | ND |
| MKs | Platelet granules | Platelet membrane | Interacts with | Effect on platelet function | |
|---|---|---|---|---|---|
| ADAM10 | Yes | ND | Yes | Tetraspanins | Release of GPVI, APP |
| ADAM17 | Yes | ND | Yes | Release of GPIbα, GPV, Sema4D, APP, sCD40L? | |
| MMP-1 | Yes | Yes | Yes | αIIbβ3, PAR-1 | Accelerates thrombus formation |
| α2β1 | Primes platelets, cleaves PAR-1, activates platelet signaling | ||||
| MMP-2 | Yes | Yes | Yes | GPIb-IX-V | Increase thrombus formation, cleaves talin |
| αIIbβ3 | Cleaves PAR-1; release of sCD40L? | ||||
| MMP-3 | Yes | Unknown | |||
| MMP-9 | Yes | Yes/No | Yes | Decreased activation; reduced Ca2+ mobilization | |
| Increased thrombus area in MMP-9−\− mice | |||||
| MMP-13 | GPVI, αIIbβ3 | Impaired platelet aggregation to low dose collagen, CRP | |||
| Reduced thrombus formation | |||||
| MMP-14 | Yes | Yes | MMP-2 | Inhibits thrombus growth and stability | |
| TIMP-2 | |||||
| TIMP-1 | Yes | Yes | Inhibits platelet ADAMs? | ||
| TIMP-2 | Yes | Yes | MMP-14 | Synthesized de novo upon activation; inhibits all MMPs | |
| TIMP-3 | Yes | Inhibits platelet ADAMs? | |||
| TIMP-4 | ND |
Location of metalloproteinases and examples of their effects on platelet function. ? indicates proposed function that needs to be experimentally determined.
CRP, collagen-related peptide; MK, megakaryocyte; ND, not detected; PAR, protease-active receptor; Yes/No, conflicting reports.
Platelet receptor shedding in mice that have platelet-specific deficiency in 1 or both of ADAM10 or ADAM17 demonstrate that murine GPVI shedding is differentially regulated. GPVI shedding triggered by calmodulin inhibitors was specifically mediated by ADAM10, however, GPVI release was mediated by ADAM17 when shedding is induced through mitochondrial damage. Although surface levels of GPVI and GPIbα were significantly increased in ADAM10/ADAM17-deficient platelets, antibody-mediated GPVI shedding still occurred, suggesting that a third possibly plasma-resident protease could cleave GPVI in vivo.64 The identity of this protease is not yet known.
Matrix metalloproteinases
The MMPs also have significant roles in platelet biology109-112 and have been reviewed recently.113,114 MMPs share many structural features with ADAM proteins, however, most MMPs do not contain transmembrane domains and are secreted from cells. MMPs are responsible for the degradation of collagen and other extracellular matrix proteins important for cell movement and inflammation.115 Megakaryocytes and platelets secrete a number of MMPs,114,116,117 which contribute to wound-healing processes and tissue remodeling. Platelet-associated MMP-1 and MMP-2 activate, and MMP-9, MMP-13, and MMP-14 inhibit, collagen-dependent platelet activation and thrombus formation possibly by interfering with GPVI and αIIbβ3 processes.111,112 At the platelet surface, both MMP-1118 and MMP-2114 can control platelet receptor signaling by cleaving protease-activated receptor 1 (PAR1) at extracellular sites different from the thrombin-cleavage site and thus selectively initiate some of the signaling pathways normally activated by full PAR1 agonism. Although direct evidence of MMP-mediated cleavage of platelet receptors is modest, it is worth noting that MMP and ADAM proteins are often capable of cleaving the same substrate within metalloproteinase-sensitive cleavage sites. This built-in redundancy may explain the maintenance of normal thrombotic responses and lack of hemostatic phenotypes in mice deficient in 1 or more metalloproteinases.
Regulation of vascular metalloproteinase activity
Substrate availability
Regulation of ADAM proteolytic specificity does not strongly rely on the availability of a signature substrate amino acid sequence. There is a wide substrate repertoire that can be targeted often by both ADAM10 and ADAM17, as well as other ADAM family members and little is understood about their cleavage site specificity. Further control and specificity is likely to be achieved via additional noncatalytic interactions of the substrate with the cysteine-rich domain of the ADAM to position the substrate for effective cleavage at the membrane.107,108 In this way, spatial information including the density of substrate can impact on the rate at which ADAM-mediated shedding can occur. The ADAM17 cleavage site within GPIbα has been shown to lie within a mechanosensory domain, which may protect it from untimely proteolysis. In the case of GPVI, the stability of this receptor on circulating inactive platelets that contain active ADAM10 may also be protected within a cryptic domain that is revealed when ligand binds, or membrane is perturbed by elevated fluid shear stress.119 Notably, ADAM10 itself can be regulated via intramembrane proteolysis in nucleated cells,120 however, there is no evidence yet that this form of regulation occurs in platelets.
Tissue inhibitors of metalloproteinase
As with other metalloproteinases, ADAMs are inhibited by members of the tissue inhibitors of metalloproteinase (TIMP) family, a group of small, secreted proteins with structurally and functionally distinct N- and C-terminal domains.121,122 ADAM10 is primarily targeted by TIMP-1 and ADAM17 by TIMP-3.123 The N termini of these inhibitors engage with the proteolytic domain of the metalloproteinase in a stoichiometric ratio of 1:1, displacing the divalent cation and blocking catalytic activity.114,124 Although other TIMPs circulate in plasma, TIMP-3 favors association with the extracellular matrix by binding to glycosaminoglycans.125 TIMPs are expressed by many cell types including endothelial and smooth muscle cells as well as platelets.126,127 The role of TIMPs in regulation of sheddases involved in platelet receptor modulation remains unstudied, however, as levels of GPVI are relatively stable on resting platelets,26 despite the presence of active ADAM10, this suggests a role for plasma and platelet-associated TIMPs or other inhibitors in preventing cleavage and stabilizing GPVI on circulating platelets.
Rhomboid proteins
The stability, trafficking, and activity of ADAM17 can also be regulated intracellularly by a specialized proteolytically inactive subset of the rhomboid family of intramembrane proteases.128 The inactive rhomboids (iRhoms) contain 7 transmembrane regions, and iRhom-2 has been shown to specifically modulate ADAM17 activity, most probably via interaction between the iRhom-2 N-terminal fragment and the catalytic region of ADAM17. Although analysis of iRhoms in platelet receptor shedding has been limited to date, both human and mouse platelets contain iRhom-2,129,130 therefore, it will be of great interest to assess the role of iRhoms on platelet receptor proteolysis by ADAM17 in future studies.
Tetraspanins
At the membrane surface, ADAM activity is regulated by tetraspanins.122 This is a family of 33 member proteins, each with 4 transmembrane domains and an ability to associate at the cell surface both with one another and with nontetraspanin integral proteins to organize a network of tetraspanin-enriched microdomains.131 Along with exercising a level of control of platelet integrin αIIbβ3,132 platelet tetraspanins principally from the TspanC8 subfamily interact with the disintegrin and cysteine-rich domains of ADAM10103,133,134 and control substrate selectivity of ADAM10 depending on which tetraspanin is associated with the metalloproteinase. Importantly, overexpression of Tspan14 (found in both human and murine platelets) but not other TspanC8 members, suppressed ADAM10-shedding activity.133 Disrupting the Tspan14-ADAM10 interaction may be a way to selectively modulate platelet ADAM10 activity for therapeutic benefit in people at risk of thrombosis.
Consequences of platelet receptor shedding
Ectodomain shedding is a ubiquitous mechanism to trigger rapid and irreversible downregulation of receptor expression. Changes of receptor expression levels over time will lead to decreased surface density and decreased ligand binding and to receptor cross linking and signaling; they may also possibly influence platelet aging or clearance (Figure 2). The ectodomain fragments may also be functional and/or act as potential platelet-specific biomarkers of platelet activation and risk of bleeding/thrombosis.
Clinically, abnormally low platelet receptor density may be associated with increased susceptibility to bleeding. The bleeding phenotype observed in people afflicted with Bernard-Soulier syndrome (absent or dysfunctional GPIb-IX-V)135,136 or in immune thrombocytopenia patients lacking GPVI due to autoantibody-mediated shedding of GPVI61,137,138 underscores the importance of these adhesion receptors in normal platelet function. Humans and mice that are deficient in GPIbα display a macrothrombocytopenia and bleeding symptoms or phenotypes that are more significant than would be expected for the platelet count. A link between the level of platelet desialylation and platelet lifespan has been described,139,140 however, recent work in GPIbα-deficient mice has shown that the N-terminal portion of GPIbα is clearly required for normal production of thrombopoietin (TPO) and that this requirement was independent of the level of platelet desialylation.141 Immobilized GPIbα ectodomain was sufficient to increase transcription of TPO messenger RNA in hepatic cells in vitro. It will be of interest to determine whether the shed fragment of GPIbα (glycocalicin) is able to trigger hepatic TPO production in vivo. Nonetheless, the GPIbα ectodomain, independent of other receptors and platelet desialylation, is a prerequisite for hepatic TPO production, and metalloproteolysis of GPIbα is likely to influence the rate and extent of TPO production.
The ADAM-mediated proteolytic release of platelet receptors may be informative in our understanding of the pathology underpinning disease processes137 and useful in stratification of thrombotic and bleeding risk142-144 and other outcomes in patients.59,145-147 In this regard, sGPVI may be a useful plasma marker of platelet activation because GPVI is platelet/megakaryocyte-specific and stable on resting platelets. Laboratory data indicate plasma sGPVI is not influenced by age or sex. GPVI shedding is a measurable consequence of ligand-mediated activation of human platelets leading to ITAM signaling, or exposure to antiplatelet autoantibodies137,148 or elevated fluid shear stress143 or with stenotic coronary vessels with altered blood rheology.60 Elevated sGPVI levels precede the occurrence of thrombotic diseases including stroke and stable angina pectoris,144,149 and onset of sepsis with fibrin exposure, and was a predictor of mortality in injured patients in intensive care.59 Elevated levels of sGPVI and loss of surface GPVI and GPIbα have been detected in patients receiving mechanocirculatory support142 and may help evaluate bleeding risk in heart failure patients in receipt of left ventricular assist devices,143 particularly when combined with other clinical markers and scoring systems.
Concluding remarks
The primary platelet adhesion receptors play a major role not only in platelet hemostatic function but also in coordinate platelet roles in inflammation, infection, and coagulopathy. Homeostatic metalloproteolysis of platelet adhesion receptors GPIbα and GPVI may be a means to control platelet responsiveness, however, the regulation of platelet proteins within seconds to minutes of platelet activation has clear ramifications for a normal hemostatic response. Dysregulated shedding, however, could significantly affect the initiation and the propagation of a thrombus, and also affect platelet interactions within the vasculature, with leukocytes, the endothelium, and with pathogens. Shedding of platelet receptors is irreversible, and platelet receptor expression cannot be restored until new platelets are produced and released into the circulation (days to weeks). Because ADAMs have a large number of substrates, this contributes to the complexity surrounding the therapeutic targeting of ADAM activity. Direct inhibition of a particular ADAM is likely to disrupt the release of a broad spectrum of biologically active fragments, however, a level of specificity may be obtained by focusing on protection of the substrate cleavage site, using reagents that can be targeted to the platelet surface and so modulate platelet ADAM activity specifically.
Acknowledgments
This work was supported by the National Health and Medical Research Council of Australia, the National Blood Authority of Australia, and ACT Health.
Authorship
Contribution: S.J.M., R.K.A., and E.E.G. all cowrote and reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Elizabeth E. Gardiner, ACRF Department Cancer Biology and Therapeutics, John Curtin School of Medical Research, The Australian National University, Building 131, Garran Rd, Acton, ACT 2601 Australia; e-mail: elizabeth.gardiner@anu.edu.au.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal