

In this issue of Blood, show that platelet factor 4 (PF4)/heparin complexes trigger complement activation (classical pathway) through naturally occurring, polyreactive immunoglobulin M (IgM). This phenomenon, which correlates with plasma IgM levels, varies widely among normal individuals, potentially accounting for heterogeneity of anti-PF4/heparin immune responses among heparin-treated patients.1
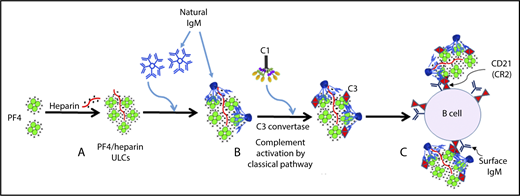
The mechanism of complement activation by PF4/heparin complexes includes the following steps: (A) heparin (polyanion) forms ULCs with PF4 (cation); (B) polyreactive natural IgM from plasma binds to PF4/heparin ULCs, and conformational changes in IgM result in binding of C1q, which leads to activation of the classical pathway of complement activation; and (C) IgM and complement-coated PF4/heparin complexes bind to B cells via complement receptor 2. See Figure 7 in the article by Khandelwal et al that begins on page 2431.
The mechanism of complement activation by PF4/heparin complexes includes the following steps: (A) heparin (polyanion) forms ULCs with PF4 (cation); (B) polyreactive natural IgM from plasma binds to PF4/heparin ULCs, and conformational changes in IgM result in binding of C1q, which leads to activation of the classical pathway of complement activation; and (C) IgM and complement-coated PF4/heparin complexes bind to B cells via complement receptor 2. See Figure 7 in the article by Khandelwal et al that begins on page 2431.
Two years ago, in a plenary paper in Blood, Khandelwal and coworkers2 reported that more than 90% of B cells obtained from normal individuals bind ultralarge complexes (ULCs) of PF4/heparin. This process somehow triggered complement activation, because PF4/heparin/complement C3 complexes were shown to bind to B cells via CD21 (ie, the B-cell complement receptor type 2 [CR2]). But exactly how this phenomenon occurs, presumably without invoking specific (cognate) antigen recognition, remained unclear.
During the development of a capture immunoassay aimed to measure complement activation in human plasma caused by PF4/heparin complexes, the authors noted marked heterogeneity in C3c generation among plasmas obtained from different healthy donors. Moreover, the amount of C3c generation seemed to be stable over time among the different donors. The authors now report the explanation for this heterogeneity in complement activation.
Khandelwal et al found that the plasma IgM levels of individuals correlate closely with complement activation triggered by PF4/heparin complexes. In contrast, correlations were not found with plasma IgG or IgA levels. By augmenting polyclonal IgM levels, C3c generation could be increased, whereas decreasing the amount of IgM had the opposite effect. In contrast, addition of polyclonal IgG or most IgM monoclonal antibodies had no effect (see below for a discussion of an IgM monoclonal antibody exception that proved illuminating). In experiments that evaluated the effects of using differing concentrations of IgM and PF4/heparin complexes, the authors concluded that complement activation by PF4/heparin complexes was indeed dependent on polyclonal IgM.
The authors then demonstrated that it is natural IgM, not immune IgM, that mediates these effects. Natural IgM refers to circulating IgM that has arisen without prior immune exposure; some natural IgM binds to recognized antigens (eg, blood groups A and B antigens and possibly PF4/heparin complexes). Natural IgM recognition of PF4/heparin complexes explains how plasma obtained from individuals who have never been exposed to heparin can achieve these IgM-mediated complement-activating effects. Interestingly, the authors also found similar complement-activating effects by protamine/heparin complexes, suggesting that these ULCs are also recognized by natural IgM. (Clinical relevance includes the fact that coadministration of protamine and heparin can cause an adverse drug effect that mimics heparin-induced thrombocytopenia [HIT].3 ) Evidence presented by the authors pointing to a role of natural IgM in these processes was demonstrated by showing similar complement-activating effects triggered by PF4/heparin (or by protamine/heparin), by cord blood (which is enriched in natural IgM), or by a monoclonal IgM (2E4) that possesses broad specificities analogous to those found in natural IgM. Finally, the authors found that an anti-C1q monoclonal antibody inhibited complement activation by IgM, thus showing that these effects are mediated by the classical complement pathway.
When incubating whole blood with PF4/heparin complexes, the authors found that complement, PF4/heparin, and IgM co-localized on the surfaces of B cells. Of clinical relevance, similar findings were seen when B cells were examined after patients had received heparin. A summary of the model proposed by these investigators (see figure) is that administration of heparin leads to formation of PF4/heparin ULCs that are recognized by natural IgM; the resulting PF4/heparin/IgM complexes trigger activation of the classical complement system, ultimately leading to formation of PF4/heparin/IgM/C3 complexes that bind to B cells via CD21.
These studies are consistent with observations made by other research groups that probe the fundamental immunobiology of the HIT immune response. For example, Zheng et al4 observed that 0.1% to 1% of circulating B cells generate anti-PF4/heparin antibodies when stimulated by certain molecules (eg, cytosine phosphatidyl guanine [CpG] oligodeoxynucleotides) known to trigger polyclonal B-cell activation. Krauel et al5 found that B cells from normal individuals (eg, from infants and from cord blood) are able to form anti-PF4/heparin antibodies after in vitro stimulation (eg, with CpG). These researchers suggested that the IgM antibody response to PF4/heparin complexes belongs to the innate immune response repertoire.
Why are these new studies reported by Khandelwal et al important? First, they elucidate the mechanisms by which PF4/heparin complexes are able to bind to most circulating B cells, which potentially helps explain how heparin can so commonly trigger an anti-PF4/heparin immune response. Second, difference in IgM levels among normal individuals could represent a stable biomarker of risk for explaining (and perhaps helping to predict) the HIT immune response (a testable hypothesis). Third, through better understanding of the HIT immune response, the likelihood of identifying some way to interrupt it increase and so does our understanding of the myriad and complex linkages between innate and adaptive immunity.
Conflict-of-interest disclosure: T.E.W. received royalties from Informa (Taylor & Francis) and lecture honoraria from Instrumentation Laboratory; provided consulting services to and/or received research funding from Aspen Global, Instrumentation Laboratory, Octapharma, and W.L. Gore; and provided expert witness testimony relating to HIT and non-HIT thrombocytopenic and coagulopathic disorders.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal