

In this issue of Blood, identified that defective activation of thrombin-activatable fibrinolysis inhibitor (TAFI) drives joint bleeding in congenital but not acquired hemophilia A.1
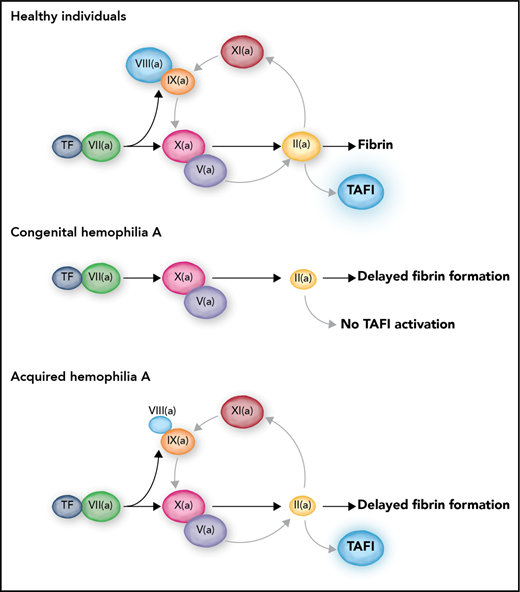
In healthy individuals, TF-induced coagulation provides rapid clot formation followed by a secondary burst of thrombin generation via FXI. This secondary thrombin burst facilitates TAFI activation, which results in protection of the clot against premature lysis. In severe congenital hemophilia, the complete absence of FVIII leads to a triple defect: a delayed clot formation, an absent FXI-mediated secondary thrombin burst, and a consequently absent activation of TAFI. In contrast, acquired hemophilia is only a single coagulation defect with delayed clot formation but preserved secondary thrombin generation and TAFI activation. Professional illustration by Somersault18:24.
In healthy individuals, TF-induced coagulation provides rapid clot formation followed by a secondary burst of thrombin generation via FXI. This secondary thrombin burst facilitates TAFI activation, which results in protection of the clot against premature lysis. In severe congenital hemophilia, the complete absence of FVIII leads to a triple defect: a delayed clot formation, an absent FXI-mediated secondary thrombin burst, and a consequently absent activation of TAFI. In contrast, acquired hemophilia is only a single coagulation defect with delayed clot formation but preserved secondary thrombin generation and TAFI activation. Professional illustration by Somersault18:24.
Hemophilia A and B are severe bleeding disorders caused by a congenital or acquired defect in coagulation factor VIII (FVIII) or FIX. Severe congenital hemophilia is characterized by spontaneous joint and muscle bleeds and bleeding after invasive procedures. The bleeding phenotype in patients with acquired hemophilia is notably different, and joint bleeds, which are common in the congenital variant, are rare in acquired hemophilia.
The question of why hemophiliacs bleed is somewhat puzzling because activation of coagulation can proceed independently of the FVIII/FIX (cofactor and enzyme) duo. Tissue factor (TF)–induced coagulation leads to activation of both FIX and FX (see figure), but it can proceed efficiently in a test tube in the absence of FIX via direct activation of FX by the TF-FVIIa complex. Indeed, the prothrombin time is not abnormal in patients with hemophilia, showing that, at least in an in vitro plasma system, FVIII and FIX are not absolutely required for clot formation. However, clot formation becomes dependent on FIX at lower concentrations of TF, which likely explains why hemophiliacs bleed predominantly in tissues with a low local TF density. In addition, FIXa is required for translocation of the coagulation reaction from the initiating TF-bearing cell to the activated platelet, and this transfer is required for large-scale thrombin generation.2 Finally, amplification of coagulation proceeds not only via TF-induced activation of FIX but also via thrombin-mediated activation of FXI, which in turn activates FIX.3
The FVIII/FIX duo is thus responsible for propagation of coagulation leading to a thrombin burst. Because the majority of thrombin is generated after formation of the fibrin clot, the next question is why all this thrombin is required for physiological hemostasis and why an absence of this thrombin burst leads to bleeding.4 One of the explanations for the requirement of large amounts of thrombin regards the activation of TAFI by thrombin. In vitro studies published in the 1990s and early 2000s have shown accelerated fibrinolysis in hemophilia and FXI deficiency caused by absent activation of TAFI as a result of substantially decreased generation of thrombin during the time after clot formation.5-7 It was demonstrated that minute amounts of FVIII7 or therapeutic doses of recombinant FVIIa8 could restore TAFI activation and normalize fibrinolytic potential in vitro. These results led to the hypothesis that hemophilia is a triple hemostatic defect characterized by (1) delayed clot formation by decreased thrombin generation at low TF concentration, (2) a decreased secondary burst of thrombin generation by defective FXI-mediated amplification of coagulation, and (3) defective downregulation of fibrinolysis by lack of TAFI activation. This model may be questioned because it assumes that TAFI activation is dependent on high concentrations of thrombin, whereas the thrombin-thrombomodulin complex is a much more efficient activator of TAFI than thrombin alone.9 Indeed, in vitro, thrombomodulin normalizes TAFI activation and downregulation of fibrinolysis in the absence of enhanced thrombin generation.10
By using a mouse model of joint bleeding, Wyseure et al provided convincing evidence that the lack of TAFI activation drives hemophilic joint bleeding and that, in this setting, thrombin (and not the thrombin-thrombomodulin complex) is the TAFI activator. In addition, the authors demonstrate that the residual amounts of functional FVIII in acquired hemophilia allow for activation of TAFI, thereby limiting the joint bleed. In acquired hemophilia, in which inhibitory antibodies against FVIII impair clot formation, the type II nature of the inhibitor results in an incomplete inhibition of FVIII activity, and because of this residual FVIII acquired hemophilia is a single, rather than a triple, hemostatic defect (see figure). A final salient finding of the study by Wyseure et al is the observation of vascular bed–specific fibrinolysis. They demonstrated that hemophilic joint bleeding is caused by defective regulation of urokinase-type plasminogen activator–induced fibrinolysis by TAFI, but that tail bleeding is driven by tissue-type plasminogen activator with no role for TAFI in controlling bleeding.
Their study provides the first in vivo evidence for a role of TAFI in hemophilic bleeding (a link that was suggested more than 20 years ago5 ) and provides an explanation for the absence of joint bleeds in acquired hemophilia. In addition, the study provides clinically relevant information because it argues against using antifibrinolytic tranexamic acid in the management of joint bleeds because tranexamic acid worsens rather than inhibits urokinase-type plasminogen activator–induced fibrinolysis.
The data presented by Wyseure et al are important because they increase ourknowledge of the physiological role of TAFI and provide important clinical clues. What the study does not deliver is an explanation for the observation that in contrast to congenital hemophilia in which bleeding severity is proportional to residual FVIII levels, the severity of acquired hemophilia seems unrelated to residual FVIII activity. Perhaps the role of TAFI is unique to the joint, and additional studies will be required to assess the contribution of TAFI to bleeding in other vascular beds. For example, it has been suggested that TAFI also contributes to bleeding in tissues with a high fibrinolytic activity such as the urinary tract, nose, oral cavity, and tonsils, but no direct in vivo evidence for this has been provided. Notwithstanding these limitations, the Wyseure et al study provides exciting new insight into the physiological role of TAFI and its contribution to hemophilic bleeding. It also suggests that alternative antifibrinolytic strategies in the management of hemophilic joint bleeds deserve clinical exploration.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal