

To the editor:
In a recent issue of Blood, O’Brien et al1 describe the development of an ex vivo model of Diamond-Blackfan anemia (DBA) using cultured peripheral blood CD34+ progenitor cells obtained from DBA patients. Although many useful models of DBA exist (including animal models,2,3 pluripotent stem cells,4,5 and RNA interference in primary human hematopoietic cells6,7 ), ex vivo models7 are particularly exciting given their immediate proximity to and development from human patients. The authors use their ex vivo model to compare gene expression patterns between sorted populations of erythroid cells at day 14 of culture that were derived from cases with DBA due to ribosomal protein (RP), GATA1, or unknown mutations to cells derived from unaffected healthy individuals.8 Between these groups, they find differences in the expression of genes involved in heme biosynthesis, translational regulation, and targets of the hematopoietic transcription factor GATA1. These results contrast with previous studies on the mechanisms underlying DBA6,9,10 (detailed comparison in Table 1). For example, in erythroid cells from DBA patients, ALAS2 transcripts were increased in the O’Brien et al study, remained unchanged in the Ludwig et al study, and were decreased in the Yang et al study, a result that has implications for the contribution of potential free heme toxicity to the severity of the anemia. Similarly, GATA1 target genes were increased in the O’Brien et al study and decreased in the Ludwig et al study, a finding that has implications for the molecular pathogenesis of DBA. Even though some of the discrepant results from these 3 studies could potentially be explained by variation in tissue source, the use of cell culture, or differences in stages of differentiation, the authors suggest a substantial revision for the underlying pathophysiology of this disorder. As the potential impact of these surprising findings is considerable for the field, we reanalyzed the raw data produced in this study in an attempt to gain further insight. Our reanalysis suggests that the major findings from this study can be attributed to confounding that was not adjusted for in the study design.11,12 We discuss this and other issues with the analysis in greater detail in the following paragraphs (reproducible R code, data, additional analyses, and additional commentary are available in Ulirsch13 ).
Comparison of gene expression results from studies of primary erythroid cells from DBA patients
| O’Brien et al study1,*,† | Yang et al study9 | Ludwig et al study6,* | |
|---|---|---|---|
| Sample type | CD34+ peripheral blood cultured for 14 d | Bone marrow | Bone marrow |
| Sample origin | DBA (RP), DBA (GATA1), DBA (I), control | DBA (RP), control | DBA (RP), control |
| Culture | 14 d | Multiple, up to 17 d | None |
| Enrichment | CD44+/CD235a−, CD44+/CD235a+ | CD71+CD36−CD235a−, CD71+CD36+CD235a+, CD36+CD235a+, CD36−CD235a+ | CD34+CD71+CD45− |
| Platform | Affymetrix human gene 2.0 ST | RT-qPCR of select targets | Affymetrix U133A |
| ALAS2 mRNA | Increased‡ | Decreased§ | No change |
| GATA1 mRNA | No change | Not measured | No change |
| β-globin mRNA | Increased‡ | Decreased§ | No change |
| GATA1 targets | Increased | N/A | Decreased |
| Heme biosynthesis | Increased | N/A | Decreased |
| Ribosome biogenesis | Decreased | N/A | Decreased |
| O’Brien et al study1,*,† | Yang et al study9 | Ludwig et al study6,* | |
|---|---|---|---|
| Sample type | CD34+ peripheral blood cultured for 14 d | Bone marrow | Bone marrow |
| Sample origin | DBA (RP), DBA (GATA1), DBA (I), control | DBA (RP), control | DBA (RP), control |
| Culture | 14 d | Multiple, up to 17 d | None |
| Enrichment | CD44+/CD235a−, CD44+/CD235a+ | CD71+CD36−CD235a−, CD71+CD36+CD235a+, CD36+CD235a+, CD36−CD235a+ | CD34+CD71+CD45− |
| Platform | Affymetrix human gene 2.0 ST | RT-qPCR of select targets | Affymetrix U133A |
| ALAS2 mRNA | Increased‡ | Decreased§ | No change |
| GATA1 mRNA | No change | Not measured | No change |
| β-globin mRNA | Increased‡ | Decreased§ | No change |
| GATA1 targets | Increased | N/A | Decreased |
| Heme biosynthesis | Increased | N/A | Decreased |
| Ribosome biogenesis | Decreased | N/A | Decreased |
mRNA, messenger RNA; N/A, not applicable; RT-qPCR, reverse transcription–quantitative polymerase chain reaction.
Gene expression comparisons are for individually SCAN- or RMA-normalized data sets.
Comparing DBA (RP) or (I) vs control.
P = .0997 and .0704, respectively: ALAS2 is in the top 1% of differentially expressed genes (based upon log2 fold change > 0.5 and P < .1) and in conflict with previous report; see analysis in Ulirsch12 for full details.
Observed across all maturation stages in culture.
By definition, an association between an exposure and an outcome is “confounded” when a third variable is associated with both (1) the exposure (in the current study) and (2) the outcome (independently) but is not simply a causal intermediate between the exposure and the outcome11 (the prototypical example of a confounded association is where coffee drinking was found to be positively associated with mortality, but after accounting for smoking habits, a confounding variable, coffee drinking was in fact protective14 ). After contemplating the unexpected directionality of the gene set enrichments (GSEs)15 observed by O’Brien et al, we wondered whether, despite the fact that the authors sorted for presumed stage-matched populations using surface marker expression (CD44+CD235a− and CD44+CD235a+), cell populations were still imperfectly matched between DBA and control samples and whether this resulted in residually confounded associations with the primary gene sets (Figure 1A).

Evidence for confounding in microarray analysis of DBA models. (A) Diagram of hypothetical relationships between exposure, outcome, and putative confounding variable in the O’Brien et al study. (B) Deconvolution of early (CD235a−) and late (CD235a+) erythroid maturation stages by CIBERSORT, a support vector regression approach, in O’Brien et al samples based upon normal erythroid maturation from GSE22552 (P = .000057 from likelihood ratio test). Error bars represent standard errors. (C) Although similarly sorted for CD235a−, DBA samples are composed of different mixtures of maturation stages than unaffected control samples. DBA due to RP or indeterminate (RP/I) samples are on average more mature (P = .017 from likelihood ratio test), whereas DBA due to GATA1 samples are less mature (P = .012 from likelihood ratio test). Error bars represent standard errors. (D) Kernel density plots for heme biosynthesis, ribosome biogenesis, and curated (cur.) GATA1 target genes. Heme biosynthesis and ribosome biogenesis are significantly associated with erythroid maturation stage (P < 10−10 for both by Kruskal-Wallis), whereas curated GATA1 target genes are only significant by more sensitive pairwise GSE analysis tests.13 Ribosome biogenesis was chosen as a more general measurement of “translational machinery” because small nucleolar RNAs were not measured in all microarrays. (E) Similar to panel D, except for the 3 groups investigated in O’Brien et al. Differences between genotype (RP/I, GATA1, or unaffected) are much smaller than between stages (Kruskal-Wallis P > .5 for all comparisons, but significant by pairwise GSE analysis13 ). (F) Similar GSE analysis to that reported by O’Brien et al is shown. Synthetic stage-matched normal samples were created using the estimate from panel B of the percentage of each erythroid stage present. GSE analysis indicates that these synthetic normals have equivalent or stronger GSE results when compared directly to the original samples. Black bars indicate genes that are ranked according to expression differences between DBA (RP/I) and unaffected controls and normalized enrichment scores (NES) are reported. All data presented in this figure were RMA-normalized (see analysis in Ulirsch13 for SCAN-normalized data). CFU-E, colony-forming unit erythroid; exp., experimental; exprs., expression; Int-E, intermediate erythroblast; late-E, late erythroblast; pheno, phenotype; Pro-E, pro-erythroblast.
Evidence for confounding in microarray analysis of DBA models. (A) Diagram of hypothetical relationships between exposure, outcome, and putative confounding variable in the O’Brien et al study. (B) Deconvolution of early (CD235a−) and late (CD235a+) erythroid maturation stages by CIBERSORT, a support vector regression approach, in O’Brien et al samples based upon normal erythroid maturation from GSE22552 (P = .000057 from likelihood ratio test). Error bars represent standard errors. (C) Although similarly sorted for CD235a−, DBA samples are composed of different mixtures of maturation stages than unaffected control samples. DBA due to RP or indeterminate (RP/I) samples are on average more mature (P = .017 from likelihood ratio test), whereas DBA due to GATA1 samples are less mature (P = .012 from likelihood ratio test). Error bars represent standard errors. (D) Kernel density plots for heme biosynthesis, ribosome biogenesis, and curated (cur.) GATA1 target genes. Heme biosynthesis and ribosome biogenesis are significantly associated with erythroid maturation stage (P < 10−10 for both by Kruskal-Wallis), whereas curated GATA1 target genes are only significant by more sensitive pairwise GSE analysis tests.13 Ribosome biogenesis was chosen as a more general measurement of “translational machinery” because small nucleolar RNAs were not measured in all microarrays. (E) Similar to panel D, except for the 3 groups investigated in O’Brien et al. Differences between genotype (RP/I, GATA1, or unaffected) are much smaller than between stages (Kruskal-Wallis P > .5 for all comparisons, but significant by pairwise GSE analysis13 ). (F) Similar GSE analysis to that reported by O’Brien et al is shown. Synthetic stage-matched normal samples were created using the estimate from panel B of the percentage of each erythroid stage present. GSE analysis indicates that these synthetic normals have equivalent or stronger GSE results when compared directly to the original samples. Black bars indicate genes that are ranked according to expression differences between DBA (RP/I) and unaffected controls and normalized enrichment scores (NES) are reported. All data presented in this figure were RMA-normalized (see analysis in Ulirsch13 for SCAN-normalized data). CFU-E, colony-forming unit erythroid; exp., experimental; exprs., expression; Int-E, intermediate erythroblast; late-E, late erythroblast; pheno, phenotype; Pro-E, pro-erythroblast.
First, we investigated whether (1) the potential confounder (maturation stage) was associated with the exposure (cells cultured from a DBA case or healthy control), focusing on the CD44+CD235a− compartment that O’Brien et al suggested was of comparable stage composition between cases and controls. As a simple first analysis, we derived several “erythroid differentiation signatures” from independent expression data sets, used multiple approaches (robust multiarray average [RMA]16 and single-channel array normalization [SCAN]17 ) to normalize the microarrays from O’Brien et al (GSE89540, accessed through the Gene Expression Omnibus), and performed GSE analysis. We found strong evidence that maturation stage was indeed associated with DBA case status and genotype (Figure 1F top panel for erythroid maturity). To more rigorously quantify the exact stage composition of each sample, we normalized 2 independent reference data sets18,19 that assayed gene expression during “normal human erythropoiesis,” corrected for potential batch effects,20 and used the CIBERSORT21 method to robustly resolve the cell-type composition of each individual sample from the authors. To assess the efficacy of our approach, we verified that independent reference sets could robustly classify one another. For example, our approach classified CD235a− samples from O’Brien et al as primarily composed of less mature stages of erythropoiesis whereas CD235a+ samples were primarily composed of later maturation stages (Figure 1B). We then applied this approach to our main question and found significant differences in stage composition based upon DBA status or genotype (Figure 1C). Most notably, we found that after 14 days of culture, CD235a− cells derived from DBA cases with GATA1 mutations were significantly less mature. Conversely, cases with RP or indeterminate mutations were overall more mature than CD235a− cells from normal donors. Although these findings were robust to choice of normalization procedure (SCAN or RMA) and reference data set (see complete analysis in Ulirsch13 ), it is important to note that this approach relies on the assumption that terminal erythroid maturation in samples from DBA patients will occur similarly as in healthy individuals.
Next, we investigated whether (2) the putative confounder (maturation stage) was associated with the outcome (gene expression), specifically with gene sets that were highlighted by the authors. We found that heme biosynthesis genes were substantially upregulated during erythropoiesis, ribosome biogenesis genes were downregulated, and GATA1 target genes were to a lesser extent upregulated during erythropoiesis (potentially decreasing in the final stages) (Figure 1D). When compared with the differences in gene set expression reported by O’Brien et al, differences between DBA cases and healthy controls were more subtle (Figure 1E). Therefore, the GSE results of this study appear to be subject to confounding by differences in stage composition based on donor DBA status and genotype. To further illustrate this, we created a “synthetic normal” expression profile22 for each sample, defined as the estimated mixture of each maturation stage:
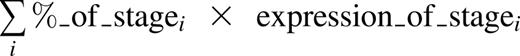
We then performed GSE and found that the stage-matched synthetic normals showed just as strong or stronger enrichments for the same gene sets, highlighting the ability of subtle differences in maturation stage to confound GSE analyses (Figure 1F; similar results comparing DBA [GATA1] to healthy controls are available in Ulirsch13 ). Thus, it is very difficult, given the currently available data, to ascertain whether these key gene sets would be overenriched, underenriched, or even unchanged between perfectly stage-matched DBA and control cells. The selection that occurs during the 14-day ex vivo culture likely enriches for cells with overall similar expression patterns, and subtle variation in differentiation therefore underlies the minor changes detectable by GSE analyses (Figure 1A).
The ability of GSE analysis to identify significant differences in imperfect biological gene sets between conditions is an enormous strength of this set of techniques. It is also an important weakness when there are true biological differences of cell-type composition that are identified by GSE analysis, but interpreted as accurate estimates of the differences between conditions. As we are certainly not immune to unintended flaws or limitations in study design, we suggest a number of approaches that can assess for and at least partially remedy confounding by cell type or maturation stage. First, multiple gene sets are usually significant at even Q < 0.01,23 and it is far more informative to consider the relative rank of the gene set of interest in comparison with all tested gene sets, especially when gene-based permutations are used instead of class permutations. Second, using orthogonal cell-type data (ie, discriminating cell surface markers, morphological characteristics, gene expression, open chromatin) to assess cell-type composition can help to identify the extent of confounding and prevent it prior to data collection. In some cases after data collection, it may actually be possible to partially correct for cell-type composition by regressing out or normalizing for these confounders,20,24 and we provide a basic example of this approach in Ulirsch,13 reaching different conclusions than those in the original report. Nevertheless, the interpretation of these results is complicated, as altered erythroid differentiation appears to be an intrinsic feature of maturing erythroid cells derived from DBA patients.7 Third, we recognize that identifying and adjusting for confounding may not always be possible, but gene expression data may have already been produced, as in this case. In such instances, GSE analysis is useful for generating hypotheses, but rigorous functional testing of these hypotheses6 and multiple lines of evidence should be evaluated prior to suggesting that any major new insights into disease pathogenesis have been gained. This is particularly important given the substantial investment in biological follow-up studies that will be based on such reports of associations from gene expression analyses.
Authorship
Acknowledgments: This work was supported by National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases grant R01DK103794 (V.G.S.) and National Institutes of Health, National Heart, Lung, and Blood Institute grant R33HL120791 (V.G.S.).
Contribution: All authors contributed to the research and edited the manuscript; J.C.U. and C.L. performed the data analysis; V.G.S. supervised all aspects of this work; J.C.U. and V.G.S. wrote the manuscript; and L.S.L., N.M., and D.G.N. provided critical comments and suggestions on this analysis.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Vijay G. Sankaran, Broad Institute of MIT and Harvard, 3 Blackfan Cir, CLS 03001, Boston, MA 02115; e-mail: sankaran@broadinstitute.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal