

Abstract
The Bruton tyrosine kinase (BTK) inhibitor, ibrutinib has produced durable remissions in chronic lymphocytic leukemia (CLL). We describe a CLL patient who progressed while receiving ibrutinib following 20 months of once daily dosing. A cysteine-to-serine amino acid replacement was identified in BTK at position 481 that disrupts the covalent, but not non-covalent, binding of ibrutinib to BTK in silico structural modeling1. The mutation was present in relapsed samples while absent in the pre-treatment and responding samples. Following the mutation, the B cell receptor (BCR) pathway was reactivated as evidenced by increased cell signaling activities and gene expression profiles. Comparing the relapsed samples with the pre-treatment and responding samples, at the cellular level, mutated CLL cells displayed higher levels of the cell proliferation marker Ki67 in vivo and higher levels of ex-vivo BrdU incorporation. Transfection of the C481S mutant construct into a sensitive lymphoma cell line rendered it much more resistant to ibrutinib treatment demonstrating the cellular impact of the mutation (see attached graph). Interestingly, the ibrutinib-resistant CLL cells remained sensitive to other BCR inhibitors such as dasatinib and SYK inhibitors. These results confirm BTK as an important pharmacologic target of ibrutinib. Further, a mechanism of resistance was revealed, and alternative therapeutic options for ibrutinib resistance were explored. (First three authors with equal contribution)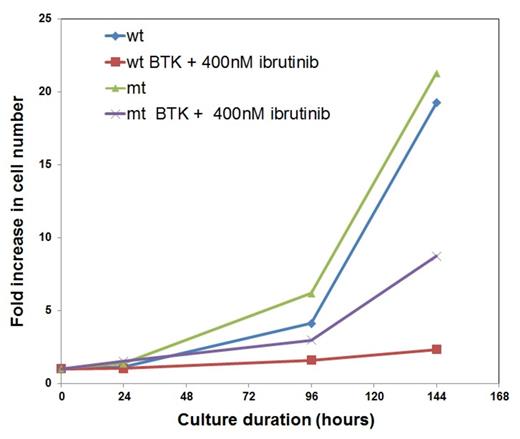

Disclosures:
Furman:Genentech: Consultancy, Speakers Bureau; GlaxoSmithKline: Consultancy, Speakers Bureau; Pharmacyclics: Consultancy; Gilead: Consultancy. Chang:Pharmacyclics: Employment, Equity Ownership.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal