

Abstract
Abstract 1687
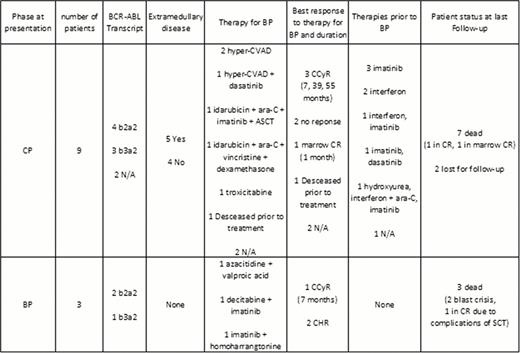
Approximately two thirds of patients with CML BP exhibit a myeloid phenotype, and one third a lymphoid phenotype. In rare occasions, patients exhibit a mixed phenotype. Biphenotypic blast phase (Bi-BP) are rare and generally portend a very unfavorable prognosis. To evaluate the incidence and outcome of Bi-BP, we reviewed 1143 patients with CML BP diagnosed and treated at M.D. Anderson Cancer Center between November 1973 and February 2012. Only Bi-BP with both myeloid and B-lymphoid differentiation was included in this analysis. Twelve (1%) patients had Bi-BP. At presentation, the median age was 62 years (range, 29–81), WBC count 12×109/L (range, 6–214), hemoglobin 10 g/dL (range, 7–14), platelet count 94×109/L (range, 41–531), peripheral blood blasts 30% (range, 0–83), and bone marrow blasts 66% (range, 0–91). Five (42%) patients had extramedullary disease involving the central nervous system (CNS; n=2), lymph nodes (n=2), and CNS, orbit and skin without bone marrow disease (n=1) (Table 1). The median time from diagnosis to transformation was 15 months (range, 0–58). Six patients expressed a b2a2 BCR-ABL1 transcript (p210), whereas 4 patients expressed b3a2 (p210) and in 2 patients such information was not available. Three patients presented initially with BP and 9 evolved to BP from chronic phase (CP). Out those 9, 6 had failed imatinib, 3 interferon, 1 dasatinib, 1 ara-C and 1 hydroxyurea prior to transformation. Median number of treatments prior to BP was 1 (range, 1 to 3). Initial therapy for Bi-BP was hyper-CVAD in 2 patients, one of them achieving complete cytogenetic remission (CCyR); hyper-CVAD and dasatinib in 1 patient, achieving a marrow CR; idarubicin, ara-C, and imatinib followed by allogeneic stem cell transplantation in 1 patient, with no response; idarubicin, ara-C, vincristine and dexamethasone in 1 patient, achieving CCyR; troxicitabine in 1 patient, achieving CCyR; combination of decitabine and imatinib in 1 patient, achieving CCyR; azacitidine and valproic acid in 1 patient with no response; and imatinib with homoharrangtonine in 1 patient with no response. Overall, 5 patients responded, to a variety of chemotherapy regimens that included a tyrosine kinase inhibitor in 2 of them. Median duration of response was 7 months (range, 1–55). One patient died during induction chemotherapy. A total of 5 patients received subsequent therapy: TKI in 3 patients and allogeneic SCT in 2 patients. Three patients developed BCR-ABL1 gene mutations: Y253H (n=1), T315I (n=1), and F317L & V299L (n=1). At last follow up, 10 patients were dead (5 due to progression) and 2 were lost to follow up. In conclusion, Bi-BP is a rare entity, which frequently exhibits an aggressive course, extramedullary disease, and high resistance to conventional chemotherapy. Therapy requires the use of chemotherapy in combination with a TKI. Nevertheless, responses are short-lived and patients should be offered allogeneic SCT or novel investigational approaches.
Summary of patients' clinical and molecular characteristics, response to treatments and outcomes
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal