

Abstract
The cellular events that lead to terminal erythroid differentiation rely on the controlled interplay of extra- and intracellular regulatory factors. Their downstream effects are highly coordinated and result in the structural/morphologic and metabolic changes that uniquely characterize a maturing red blood cell. Erythroid Krüppel-like factor (EKLF/KLF1) is one of a very small number of intrinsic transcription factors that play a major role in regulating these events. This review covers 3 major aspects of erythropoiesis in which EKLF plays crucial functions: (1) at the megakaryocyte-erythroid progenitor stage, where it is involved in erythroid lineage commitment; (2) during the global expansion of erythroid gene expression in primitive and definitive lineages, where it plays a direct role in globin switching; and (3) during the terminal maturation of red cells, where it helps control exit from the cell cycle. We conclude by describing recent studies of mammalian EKLF/KLF1 mutations that lead to altered red cell phenotypes and disease.
Introduction
Erythroid Krüppel-like factor (ELKF/KLF1)1 is the founding member of the Krüppel-like factor (KLF) family that comprises 17 different transcription factors playing critical roles in diverse biologic processes.2-4 KLFs are characterized by a highly conserved DNA-binding domain that belongs to the well-studied class of C2H2 zinc fingers.5,6
EKLF is essential for erythropoiesis.7,8 Its expression is restricted to the hematopoietic organs throughout all developmental stages, initiating at embryonic day 7.5 (E7.5) in the yolk sac, then switching to the fetal liver, and finally to the adult bone marrow and the red pulp of the spleen.9 EKLF protein is functionally present in both primitive and definitive erythroid populations.10,11
EKLF consists of 2 domains: an N-terminal proline-rich transactivation domain and a C-terminal DNA-binding domain that recognizes the consensus binding site 5′CCN CNC CCN.12 Recently, this motif has been more precisely defined and comprises the sequence 5′ CCM CRC CCN (where R represents A or G and M represents A or C), thus providing additional specificity to the “N” positions previously predicted from in vitro studies.13,14 Such a motif is found in the regulatory regions of many erythroid genes. EKLF can also bind to noncanonical consensus sites as found in the AHSP promoter; however, it is not clear whether the surrounding nucleotides or the overlapping nature of the CACC sites facilitate binding.15
An extensive description of EKLF biochemical and molecular studies are beyond the scope of this review. In brief, EKLF has been primarily characterized as a transcriptional activator, but can also exert transcriptional repression. EKLF activity is modulated by posttranslational modifications that promote protein-protein interactions (Figure 1), some of which follow from its interaction with cofactors p300/CBP. Acetylation of lysine 288 and the phosphorylation status of threonine 41 are critical for optimal transactivation activity.16-18 Acetylation of K288 in particular leads to increased association of EKLF with the chromatin remodeling SWI/SNF-related complex in vitro, yielding an open chromatin domain and transcription of the adult β-globin promoter.19-21 Integration of transcription and chromatin-modifying activities enables EKLF to play a central role in formation of the active chromatin hub across the β-like globin locus.22 Given its prominent role as an activator, it is intriguing that EKLF can also act as a repressor. For example, sumoylation of K74 is implicated in EKLF repression activity, likely by tethering components of the NuRD complex to specific DNA targets.23

Schematic diagram of the EKLF protein. Shown are N-terminal proline-rich and C-terminal (C2H2 zinc fingers; ZnF) DNA-binding domains. EKLF activity is modulated by posttranslational modifications that promote protein-protein interactions. The transactivation activity of EKLF (top) is associated with the phosphorylation status of threonine 41 and acetylation of lysine 288.16-18 Acetylated K288 is involved in interactions with the chromatin-remodeling SWI/SNF-related complex (E-RC1) through the BRG1 subunit.19-21 The repression activity of EKLF (bottom) relies on an acetylation of K302, enabling its interaction with corepressors Sin3a and HDAC1.91,92 Sumoylation of K74 serves to tether components of NuRD repression complex via interaction with Mi2β and HDAC1.23 EKLF is also a substrate for ubiquitination; however, this modification is not specific to any particular internal lysine.93
Schematic diagram of the EKLF protein. Shown are N-terminal proline-rich and C-terminal (C2H2 zinc fingers; ZnF) DNA-binding domains. EKLF activity is modulated by posttranslational modifications that promote protein-protein interactions. The transactivation activity of EKLF (top) is associated with the phosphorylation status of threonine 41 and acetylation of lysine 288.16-18 Acetylated K288 is involved in interactions with the chromatin-remodeling SWI/SNF-related complex (E-RC1) through the BRG1 subunit.19-21 The repression activity of EKLF (bottom) relies on an acetylation of K302, enabling its interaction with corepressors Sin3a and HDAC1.91,92 Sumoylation of K74 serves to tether components of NuRD repression complex via interaction with Mi2β and HDAC1.23 EKLF is also a substrate for ubiquitination; however, this modification is not specific to any particular internal lysine.93
Genetic ablation of EKLF leads to profound β-thalassemia and embryonic lethality at day E14-E15 of gestation due to defective definitive erythropoiesis partly caused by a major deficit in β-globin expression.7,8 However, restoration of β-like globin alone is not sufficient to rescue the lethal phenotype, suggesting additional deficits in the expression of other EKLF target genes.24 Recent analysis of differential gene expression between wild type (WT) and EKLF-deficient fetal liver cells,15,25,26 together with EKLF chromatin immunoprecipitation coupled with high-throughput DNA sequencing (ChIP-seq) studies,13 have expanded the target repertoire of EKLF beyond that of adult β-globin, implicating a more global regulatory role for its activity during erythropoiesis.
Dual role of EKLF in establishing lineage fate
Hematopoiesis is a relatively linear and hierarchical process in which hematopoietic stem cells undergo successive symmetric and asymmetric divisions to yield committed progenitor cells. Stem cell potency is progressively restricted as they differentiate toward mature lymphoid, myeloid, and erythroid lineages. Differentiation at each stage depends on the interplay and equilibrium of different sets of transcription factors.27 EKLF is not present in hematopoietic stem cells and is barely detectible in multipotent myeloid progenitors. However, starting with common myeloid progenitor cells and their megakaryocyte-erythroid progenitor (MEP) progeny, expression of EKLF selectively increases as cells mature toward the erythroid lineage. In sharp contrast, EKLF levels are down-regulated in those MEPs that differentiate toward megakaryopoiesis, such that bipotential differentiation of MEPs results in erythroid progenitors exhibiting an 80-fold greater level of expression of EKLF compared with that seen in megakaryocyte progenitors.28
Mouse embryonic stem cell lines with doxycycline-inducible EKLF have been used to study its role during MEP differentiation, particularly to test the effect of EKLF misregulation (gain-of-function) on the normal hematopoietic progression as the embryonic stem cells differentiate to embryoid bodies (EBs).28 Forced expression of EKLF negatively affects megakaryopoiesis, as shown by significant decreases in megakaryocytic markers (double-positive CD41/CD42b and CD41/CD42d) in EKLF-induced cultures compared with uninduced cultures. Intriguingly, this coincides with higher levels of mature CD71/Ter119 red cell markers. Morphologically, the induced erythroid population contains more differentiated basophilic and polychromatophilic erythroblasts than uninduced cultures. The timing of EKLF induction during EB differentiation is important for both megakaryocyte inhibition and erythroid lineage promotion28 (Figure 2).

Role of EKLF in erythroid lineage development. At the MEP stage, EKLF represses megakaryopoiesis while at the same time it promotes erythropoiesis. This process likely relies, at least in part, on cross-antagonizing interactions between EKLF and Fli-1, a crucial megakaryocyte transcription factor. Committed erythrocyte progenitors expand through renewal divisions that occur during fetal erythropoiesis or after hypoxic stress.66 Terminal maturation of erythrocytes occurs throughout differentiation divisions, where cells undergo many changes: size reduction, hemoglobin accumulation, decrease in overall gene expression level, nuclear condensation, and ultimate extrusion. These events are particularly dependent on the presence of, and in some cases, the level of EKLF. The inset shows the progression of erythroid development in WT and EKLF-deficient fetal liver cells as monitored by presence of the cell-surface markers CD71 and Ter119. Discrete populations of cells (R1-R5) with distinct morphologies can be distinguished, which correspond to particular stages of cells during erythropoiesis, as described in “Dual role of EKLF in establishing lineage fate.” (Used with permission from Pilon et al.34 )
Role of EKLF in erythroid lineage development. At the MEP stage, EKLF represses megakaryopoiesis while at the same time it promotes erythropoiesis. This process likely relies, at least in part, on cross-antagonizing interactions between EKLF and Fli-1, a crucial megakaryocyte transcription factor. Committed erythrocyte progenitors expand through renewal divisions that occur during fetal erythropoiesis or after hypoxic stress.66 Terminal maturation of erythrocytes occurs throughout differentiation divisions, where cells undergo many changes: size reduction, hemoglobin accumulation, decrease in overall gene expression level, nuclear condensation, and ultimate extrusion. These events are particularly dependent on the presence of, and in some cases, the level of EKLF. The inset shows the progression of erythroid development in WT and EKLF-deficient fetal liver cells as monitored by presence of the cell-surface markers CD71 and Ter119. Discrete populations of cells (R1-R5) with distinct morphologies can be distinguished, which correspond to particular stages of cells during erythropoiesis, as described in “Dual role of EKLF in establishing lineage fate.” (Used with permission from Pilon et al.34 )
Reciprocal loss-of-function analyses have been performed using primary EKLF-deficient mouse yolk sac or fetal liver cells (as sources of primitive or definitive erythropoietic cells),28-30 human cord blood progenitor cells, and mouse erythroleukemia (MEL) cells that stably express an anti-EKLF shRNA.31 In contrast to the gain-of-function studies, EKLF knockout embryos yield an increased number of circulating platelets, and E13.5 fetal liver cells from EKLF-null embryos cultured in the presence of thrombopoietin show more than a 2-fold increase in double-positive CD41/42b and CD41/42d populations compared with WT fetal liver cells. Moreover, megakaryocyte colony numbers are 3- to 4-fold greater from an unsorted or a common myeloid progenitor–sorted population when derived from EKLF-deficient cells. These colonies are more expanded in size compared with the relatively compact colonies derived from the WT cells. Therefore, EKLF-null fetal liver cells produce a greater number of megakaryocyte progenitors and their progeny than do WT cells.28 However, at the genetic level there is considerable misregulation of megakaryocyte and erythroid expression patterns resulting from loss of EKLF. Both primitive (EryP) and definitive (EryD) erythrocytes (CD71+ population) from EKLF-null embryos show abnormal expression of a range of intracellular and cell-surface proteins.29,30 For example, EKLF-null or EKLF-heterozygous EryP cells fail to express Ter119.25,30 At the same time, CD41 is aberrantly expressed on the surface of circulating E12.5-E13.5 EryP and E14.5 EryD EKLF-deficient cells, and expression of the gene for Itga2b/GpIIb is increased at least 80-fold over that measured in WT or EKLF-heterozygote EryP cells.30 mRNA levels for Fli-1, a transcription factor that plays a critical role in megakaryopoiesis, is increased 25-fold in EKLF-null EryP cells.30
Generally, the loss of EKLF leads to a failure to silence megakaryocyte-specific genes in erythroid progeny from the MEP, resulting in a partial identity crisis within maturating EryP and EryD cells. Specification to the erythroid lineage is compromised in favor of the megakaryocyte program, which is more pronounced in EryD cells.29,30 These observations suggest that EKLF regulates cell identity not only in MEP progenitors, but also throughout EryP or EryD cell maturation. Consistent with this, the EKLF knockdown MEL cell line is associated with both significant reduction of EKLF and its target genes and increase of megakaryocytic cell-surface markers and mRNA levels of Gp1b, GpIX, GpIIIa, and Fli1.31 Similarly, the expression level of erythroid marker GPA is reduced, and in parallel the number of CD41/CD42 double-positive cells are increased, in human cord blood progenitor cells treated with shRNA against EKLF. However, the timing is critical, because already-committed erythrocytes treated with shRNA do not show any reactivation of megakaryocytic genes, nor is there any biphenotypic readout.31
These studies suggest that EKLF plays a dual role in hematopoiesis (Figure 2) by promoting erythroid lineage development while at the same time repressing or attenuating the megakaryocytic program, not only in the MEP population but also continuously throughout erythroid maturation. This distinguishes EKLF from other commonly expressed transcription factors (such as Gata1 and p45-NFE2) that play critical positive roles for both megakaryocytic and erythroid lineages.
Whereas the mechanisms by which EKLF directs lineage fate decisions and differentiation programs remain incompletely understood, comparative analysis of expression arrays between WT and EKLF-deficient fetal liver cells reveal differential expression of the transcription factor Fli-1, a critical megakaryocytic activator.28 Functional cross-antagonism between Fli1 and EKLF might be involved in the commitment toward erythroid versus megakaryocytic lineages (Figure 2). EKLF and Fli-1 can physically interact in vitro and therefore may negatively affect each other's activity. Fli1 can inhibit EKLF-dependent transcription of the β-globin gene in cotransfected MEL cells, and EKLF can inhibit Fli-1–dependent transcription of the megakaryocyte GPIX gene in cotransfected COS7 cells.32 Moreover, EKLF overexpression in differentiating EBs leads to a dramatic down-regulation of Fli1 mRNA levels within 24 hours.28 EKLF also activates the BKLF repressor,33 the expression of which may be implicated in inhibition of megakaryocytic genes.29,30 In addition, Fli-1 and EKLF might compete for limiting amounts of common cofactors such as GATA-1 during formation of mutually exclusive multiprotein complexes that selectively activate either erythrocytic or megakaryocytic promoters. In support of this, EKLF knockdown is associated with a simultaneous increase of both Fli-1 and GATA-1 recruitment at megakaryocytic gene promoters.31
The sumoylation status of EKLF is critical for its effect on megakaryopoiesis. Transgenic mice designed to misexpress EKLF in the megakaryocytic progenitor compartment inhibit megakaryocyte colony formation and cellular output. However, a sumoylation-deficient mutant of EKLF, expressed in the same way, has no effect on megakaryocyte development. As a result, the posttranslational modification status of EKLF and the identity of its protein partners may be relevant to any directive EKLF role in bipotential decisions from the MEP.23
EKLF in global expression of erythropoietic genes
The differentiation status of MEP progeny committed to the erythroid lineage can be monitored by the relative expression of the cell-surface markers CD71 (transferrin receptor) and Ter119. Discrete populations of cells, enriched for a particular stage of erythropoiesis and characterized by distinct morphologies, can be separated by flow cytometry using these markers. In E13.5 WT liver cells, 5 populations can be distinguished: R1 (CD71LO Ter119NEG, early erythroid progenitor; BFU-E), R2 (CD71HI Ter119LO, erythroid progenitor; CFU-E), R3 (CD71HI Ter119HI, proerythroblast, basophilic erythroblast), R4 (CD71MID Ter119HI, polychromatic erythroblast, orthochromatic erythroblast), and R5 (CD71LO Ter119HI, reticulocytes and erythrocytes).34,35
Comparison of the CD71/Ter119 populations in fetal liver cells (E13.5) reveals that only the R1 and R2 categories are present in EKLF-deficient embryos, demonstrating that a lack of EKLF prevents the progression of erythropoiesis between the R2 and R3 stages (Figure 2). Alternatively, the Ter119 epitope might be a direct target of EKLF. In this case, the R3-R5 populations might be present but not recognizable by FACS for Ter119. Gene-expression profiling of sorted R1+R2 populations from WT and EKLF-null fetal liver cells shows dysregulation of ∼ 3000 genes (2534 are down-regulated and 665 are up-regulated in EKLF-null embryos). Perturbation of genes involved in cell-cycle and DNA replication pathways predominate.34 Consistent with a role for EKLF in erythroid progression, the level of E2F2, a transcription factor that controls S-phase entry and DNA synthesis, is significantly decreased in the EKLF-null cells to < 5% of WT. Cell-cycle profiles of sorted R1+R2 E13.5 fetal liver cells confirm impaired progression from the G1 to the S phase in erythroid progenitor and precursor cells lacking EKLF. E2F2 has been shown to be a direct target of EKLF,34,36 and EKLF binding to E2F2-regulatory elements maintains the locus in an active chromatin state. In turn, E2F2 protein transactivates other cell-cycle pathway members, allowing progression of early erythroid progenitor cells, including BFU-E and/or CFU-E, through the G1/S transition before terminal erythroid differentiation. Insufficient expression of E2F2 might be a rate-limiting factor in erythroid precursor progression and could explain, at least in part, the lack of terminally differentiated erythrocytes in EKLF-deficient fetal liver cells.34 Therefore, during erythropoiesis, EKLF's function is essential for the completion of terminal differentiation beginning at the proerythroblast stage.26,34
Initially, the lethal EKLF-null phenotype was thought to be primarily due to the loss of β-globin gene expression, but the deficit is more severe than that seen in mice with an inactivated adult β-globin gene,37 and correction of globin chain imbalance fails to completely rescue EKLF-null mice.24,38,39 These data argue for the existence of additional nonglobin defects in EKLF-null red cells. Several approaches have been undertaken to address this issue and to generate a more complete list of EKLF target genes, including comparisons between transcriptional profiles of WT versus EKLF-deficient EryD cells15,25,26 or of the B1.6 EKLF-null cell line (now called K1-ER cells) with or without EKLF-dependent rescue.25 These studies share a remarkably similar set of potential direct EKLF target genes. Many more genes are down-regulated in EKLF-null cells than are up-regulated, confirming EKLF's primary role as a transcriptional activator in vivo. In addition to the β-globin gene—the best-studied EKLF target—putative targets comprise genes essential for all categories of red blood cell function, including red cell membrane, cytoskeletal, heme biosynthetic, transmembrane proteins, transcription and cell-cycle factors, and blood group antigens. Representative examples from these categories are detailed below.
EKLF targets in definitive cells
Expression of dematin (Epb4.9), an important cytoskeletal protein required for membrane integrity and stability in erythrocytes,40 is highly dependent on EKLF, because WT fetal liver cells express an ∼20-fold greater level compared with EKLF-null cells.25,26 In addition, mRNA levels for several other membrane-associated proteins (ankyrin, β-spectrin, band 3, aquaporin, Rh30, p55, protein 4.2, and ERMAP) show significant differences in expression in EKLF-deficient versus WT fetal liver cells.15,25,26
α Hemoglobin stabilizing protein (AHSP), a small protein that binds free α-globin chains, is another EKLF target gene for which expression is virtually absent in EKLF-null fetal liver cells and which is dramatically up-regulated in K1-ER cells after EKLF rescue.25 AHSP inhibits production of reactive oxygen species from α-hemoglobin and prevents the precipitation of highly unstable, cytotoxic free α-globin chains in red cells.15,25,26,41
Heme biosynthetic pathway enzymes, such as Alas2, Pdbg, and Bzrp, are reduced in EKLF-null red cells. This may account for the defective heme synthesis and siderotic (mitochondrial) iron accumulation phenotype.25,26
EKLF-null fetal liver cells show little Ter119 expression.25,30 In addition, expression of the transferrin receptor 1 (TfR1), another transmembrane gene commonly used as an erythroid marker, is reduced.25
The Gardos channel, encoded by Kcnn4, has been implicated in cell size reduction during erythroid maturation. EKLF-null progenitors differentiated in culture do not reduce their cell size, which implicates Kcnn4 gene as a potential EKLF target.26
Expression-profiling experiments suggest that many blood group antigens are regulated by EKLF; for example, Rh-cde, Duffy, ICAM4/LW, Ermap/Scianna, glycophorin A (GPA), CD47 (an Rh antigen–associated protein), and CD59 (a GPI-anchored protein lost in paroxysmal nocturnal hemoglobinuria) are differentially expressed.25 Interestingly, most of these proteins coexist in a mega-complex that links the membrane to the underlying cytoskeleton.42 Mouse Rh-cde, GPA, CD59, CD47, and Ermap/Scianna genes all contain CACC sites in their proximal promoters that are likely to bind EKLF.43,44
EKLF targets in primitive cells
The expression of novel EKLF target genes has also been examined in E11.5 embryonic blood during primitive erythroid cell differentiation.25,26,30 There is a dramatic reduction in expression of AHSP, dematin, Mgst3, Ermap/Scianna, Acp3, the peripheral benzodiazepine receptor (Bzrp), Rh-cde, and ICAM4/LW in EKLF-null embryonic red cells. Primitive EKLF-null red cells also show defects in gene expression and surface exposure of Ter119. Loss of dematin and Ter119 might partly explain the abnormally ruffled red cell membrane. Despite this abnormal phenotype, the embryonic EKLF-deficient red cells are not significantly different in gross morphology from their WT littermates. Given that EKLF-null primitive erythrocytes appear to have a relatively normal (albeit transient) lifespan and that embryos survive until later in gestation, it is likely that other CACC-binding proteins, such as LKLF (KLF2), may partly compensate for the absence of EKLF at certain promoters of genes such as the ζ-, ey-, and bh1-globins,45 as well as nonglobin genes.25 These data implicate EKLF as an essential regulator of global erythroid gene expression in both primitive and definitive red cell populations.
Genomic EKLF occupancy of regulatory regions
To ascertain whether genes identified from the genome-wide expression profiling experiments are indeed direct EKLF transcriptional targets, EKLF ChIP-seq in primary erythroid tissue has been performed.13 At least 945 sites in the genome of E14.5 fetal liver erythroid cells are occupied by endogenous EKLF. Sixteen percent of these in vivo EKLF-binding sites occur within 1 kb of transcription start sites, but the majority (52%) are located at distances > 10 kb from any known transcription start site.13 These observations confirm that EKLF is a direct regulator of many differentially regulated genes, particularly those involved in the production of the globins, heme, and blood group antigens, and in the maintenance and integrity of the erythroid cell membrane and cytoskeleton. Intriguingly, EKLF binding is implicated in the regulation of apoptosis in red cells, being detected within the antiapoptotic, pro-survival Bcl2l1 (Bcl-X), Xiap, Rad23a, and Pim1 genes, and also paradoxically within the proapoptotic Siva1 and Ctsb genes.13,46 EKLF also occupies the Fn3k, Slc2a4, and Pigq genes, suggesting a direct role in sugar metabolism and protein modification such as glycosylation and GPI biosynthesis. EKLF occupancy within the α-globin gene cluster is detected at HS-31, HS-26, HS-21, and HS-8, and in the Hba-a2 and Hba-a1 gene promoters,13 which is consistent with earlier ChIP analyses at that locus.47
EKLF ChIP-seq peaks are found in the vicinity of the transferrin receptor (Tfrc1; CD71), the mitochondrial iron transporter mitoferrin (Slc25a37), and the Slc11a2 and Steap3 genes that encode proteins involved in iron transport and iron reduction in the endosome.13 EKLF directly regulates the Abcb10 (ABC-me) and Abcg2 genes that produce heme exporter proteins involved in the final step in the assembly of the hemoglobin molecules.13
The ChIP-seq analysis reveals that EKLF occupancy lies within the first intron or at intragenic regions of a large majority of the activated target genes and not at the promoter region. This finding supports the notion that in many cases EKLF may activate target genes at alternative erythroid-specific promoters or via intronic enhancer elements, as has been shown at the E2f2, E2f4, and p21 (Cdkn1a) genes.13,36,48 However, the global binding pattern is not static, because the categories of EKLF occupancy in erythroid progenitors are not equivalent to that seen in mature erythroblasts.49
The degree to which gene activation occurs depends on many factors in addition to the binding of this single transcription factor. EKLF and GATA1 co-occupy many sites in the erythroid genome in a distinct, nonrandom configuration.13 For example, 48% of EKLF ChIP-seq peaks have a corresponding GATA1 ChIP-seq peak within 1 kb, suggesting that the 2 transcription factors cooperate at each of these cis regulatory elements in vivo.13
Coordination of global erythroid gene expression
Recent studies establish a new gene-expression paradigm by showing that coregulated genes together with their regulatory factors preferentially cluster at specialized transcription factories.50 Such factories are associated with the active, hyperphosphorylated forms of RNA polymerase II (RNAPII S5P) to optimize efficient and coordinated transcriptional control. EKLF mediates preferential (at very high frequencies; 63%-79%) co-associations of EKLF-regulated genes at a limited number of these specialized transcription factories. Nearly all EKLF foci overlap with RNAPII-S5P foci, indicating that 10%-20% of transcription factories in mouse erythroid cells contain high levels of EKLF.50 These interactions are specifically disrupted in the absence of EKLF. Hbb, Epb4.9, and Ahsp all show markedly reduced association with transcription factories in EKLF-null erythroid cells, which is consistent with their dependence on EKLF for expression. Both intrachromosomal (Hbb-Kcnn4) and interchromosomal (Hbb-Hba; Hbb-Epb4.9) interaction frequencies were reduced in EKLF-null cells. These data indicate that EKLF is required for preferential colocalization of EKLF-regulated genes at shared transcription factories.50
The majority of EKLF-regulated genes are not absolutely dependent on EKLF for basal expression, but instead require EKLF for increased expression to attain optimal levels in the definitive erythroid lineage. The individual genes may indirectly benefit from cooperative associations in these specialized microenvironments. The preferential associations in transcription factories substantially affect higher-order chromosomal conformations and are a major driving force in tissue-specific chromosomal positioning.50,51
EKLF in globin switching
One of the main genetic signatures distinguishing primitive and definitive erythropoiesis is the expression of stage-specific hemoglobin variants. The transitions from embryonic to fetal and to adult β-like globin expression accompany a shift in the primary site of erythropoiesis from the yolk sac to the fetal liver and ultimately the bone marrow.52 Historically, the role of EKLF in enabling the final switch to adult β-globin has been the major focus of genetic, biochemical, and molecular studies since its discovery.53,54
Developmental stage–specific alterations in EKLF abundance may also help to mediate the competitive interactions of β-like globin gene family members with the far-upstream locus control region. The level of mouse EKLF increases 3-fold in definitive erythroid progenitors compared with primitive erythroid progenitors.55 Similarly, mice with a low-expressing hypomorphic EKLF allele demonstrate delayed silencing of murine embryonic globins (and transgenic human fetal globin).56
Recent studies have shown that EKLF plays a critical role in regulating the developmental switch between fetal and adult hemoglobin expression, both by direct activation of β-globin and indirect repression of γ-globin gene expression in adult erythroid progenitors via regulation of Bcl11a57 (Figure 3). For example, the hereditary persistence of fetal hemoglobin (HPFH) phenotype within a Maltese family has been mapped to a heterozygous nonsense mutation (K288X) in EKLF that leads to haploinsufficient expression. Genome-wide expression studies show that among known EKLF targets, the γ-globin repressor Bcl11a58-60 is highly down-regulated in these individuals, whereas embryonic and fetal globin are markedly up-regulated.57 Manipulated EKLF levels are directly proportional to Bcl11a levels and inversely proportional to γ-globin levels, and ChIP analyses confirm a direct EKLF occupancy within the promoter region of Bcl11a gene in both humans and mice.56,57 Consistent with these data, butyrate treatment of transgenic murine fetal liver definitive erythroid cells down-regulates levels of EKLF and Bcl11a while increasing embryonic and fetal globin expression.61

EKLF regulates globin switching. During embryonic and fetal development or in EKLF-haploinsufficient adults (left), EKLF levels are relatively low, resulting in low levels of adult β-globin and Bcl11a and high levels of γ-globin. In adults with 2 functional copies of EKLF (right), increased expression of EKLF in definitive red blood cells promotes high levels of adult β-globin and Bcl11a expression, the latter of which in turn represses γ-globin expression. (Modified from Bieker.94 )
EKLF regulates globin switching. During embryonic and fetal development or in EKLF-haploinsufficient adults (left), EKLF levels are relatively low, resulting in low levels of adult β-globin and Bcl11a and high levels of γ-globin. In adults with 2 functional copies of EKLF (right), increased expression of EKLF in definitive red blood cells promotes high levels of adult β-globin and Bcl11a expression, the latter of which in turn represses γ-globin expression. (Modified from Bieker.94 )
EKLF may contribute even more directly to β-like globin gene switching. Tagged EKLF interacts in vivo with the mouse embryonic β-like globin or the transgenic human fetal γ-globin promoter in primitive erythroid cells, and then switches to bind the mouse and human adult β-globin promoter in definitive cells.55,56 Although EKLF is not absolutely necessary for embryonic/fetal globin expression, it has quantitative effects on the levels of some of these genes.62-64 Its interaction with these promoters may help to establish their most optimal transcriptional activity before the final switch, at which point the lower affinity of EKLF for the γ-CACCC versus the β-CACCC element12 may enable a quick release and switch when the β-site becomes available. A complication is that EKLF levels are lower in the very cells (primitive) within which it is postulated to act at a suboptimal binding affinity target (the embryonic/fetal CAC site). It remains to be demonstrated whether EKLF binds the γ-globin promoter in human fetal liver cells. However, it is intriguing that treatment of primary human erythroid precursors with short-chain fatty acid derivatives leads to an increase in the occupancy of EKLF at the γ-globin promoter.65
Based on all of these observations, EKLF knockdown could be used to increase γ-globin expression in a therapeutic setting. However, such a design may compromise the expression of β-globin and other selected genes if it is even slightly too low (see “EKLF mutations related to hematologic parameters and disease”).
EKLF during red cell terminal maturation
The switch from proliferation to differentiation during the terminal stages of tissue maturation is a tightly controlled process that relies in part on transcription factor–mediated activation of cell-cycle components. In general, differentiation is associated with cell-cycle arrest. However, in the erythroid lineage, early stages of terminal differentiation are coupled to proliferation. Later stages of erythroid differentiation, from erythroid precursors to mature hemoglobinized erythrocytes, require 3-4 cell divisions, during which the cell size is reduced, gene expression levels decrease, and the nucleus condenses—and in the ultimate gene-silencing process is lost by enucleation.66,67
Divisions during erythroid maturation
As mentioned previously, EKLF, via activation of the E2f2 locus, is required for the cell-cycle progression that precedes terminal erythroid differentiation.34,36 Compared with WT cells, EKLF-null E13.5 fetal liver definitive erythroid cells show a significant increase in the percentage of cells with G0/G1 (2N) DNA content and a parallel decrease in the percentage of cells containing S-phase (> 2N) or G2/M (4N) DNA content. A similar reduction in cells with S-phase DNA content is observed in EKLF-null cells during primitive (E10.5) erythropoiesis. Therefore, expression of E2F2 is significantly reduced in both primitive and definitive erythropoiesis in EKLF-deficient embryos. However, expression of the closely related transcription factor E2F4 is affected only at E14.5 in EKLF-null fetal liver compared with WT.36 E2F2 and E2F4 are components of the Rb-E2F complex that controls S-phase entry and that are highly expressed during erythroid differentiation. A loss of either transcription factor in mice leads to macrocytic anemia, which is associated with defects in erythroid maturation.36,68,69 Cell division and reduction in cell size are tightly linked, suggesting that the major role of E2F2 and E2F4 in erythropoiesis is the promotion of cell proliferation through the direct transactivation of downstream cell-cycle genes. ChIP analysis in primary fetal liver cells shows endogenous EKLF occupancy at E2f2 and E2f4 regulatory elements located in their promoter and intronic regions, some of which exhibit strong enhancer activity.13,34,36 The appearance of enlarged red cells in the peripheral blood of E2F2-null mice is consistent with premature cell-cycle exit.
Restoration of the cell-cycle balance in EKLF-null red cells by the additional loss of Rb partially rescues S-phase entry during definitive erythropoiesis.36 However, many other aspects of erythroid development remain perturbed; for example, there is no indication of a differentiation rescue, shown by the persistent absence of erythroid cell-surface markers Ter119 and CD71.36
Cell-cycle exit at late stages of terminal erythroid maturation
The level of EKLF protein changes during erythroid development. An antiproliferative effect of EKLF is suggested by an increased derivation frequency of immortal clones from EKLF-null compared with EKLF-heterozygous fetal liver cells after J2 virus infection, indicating that EKLF discourages immortalization.70 The absence of EKLF also results in a markedly increased rate of transformation by the cooperating oncogenes v-mil-raf and v-myc. It is perhaps then not surprising that increased levels of EKLF during terminal maturation are correlated with the transition of the cell-cycle status from proliferation to differentiation.71,72 This change is mediated (at least in part) by EKLF's direct activation of the cyclin-dependent kinase (Cdk) inhibitors p18 and p21.48,73 p18 is a member of Ink4 family that inhibits cyclin D-CDK4 complexes in early G1, whereas p21 belongs to the CIP/KIP family that regulates cell-cycle transition through inhibition of cyclin E-CDK2 complexes at later stages of G1.
Analysis of terminal differentiation in G1E-ER cells after β-estradiol–induced nuclear translocation of GATA1 results in a concomitant increase in the expression of EKLF and the cell-cycle inhibitor p21; however, siRNA knockdown of EKLF under these conditions proportionately decreases the expression of p21.48 Similarly, forced overexpression of EKLF in MEL cells is directly correlated with an increase in mRNA and protein level of p21, exit from the cell cycle, and G0/G1 arrest.48 The Cdk inhibitor p18 was identified as a potential novel EKLF target gene in an expression-profiling study.73 ChIP analyses performed using erythroid cell lines confirmed EKLF occupancy at regulatory elements located in the proximal promoter and intron 1 of the p21 gene and at an upstream promoter region of the p18 gene.48,73 These data are consistent with the notion that EKLF levels directly activate p18 and p21 and help mediate cell-cycle arrest. Neither the p18 or the p21 gene is strongly occupied by EKLF in whole fetal liver nuclei. This may reflect the heterogeneity of erythroid cell differentiation states in fetal liver and possible dynamic occupancy of EKLF at different cell-cycle gene regulators as differentiation progresses.
In summary, EKLF promotes the differentiation and maturation of red cells by direct regulation of several components of the cell-cycle machinery. However, the complex set of events that occur at the final erythroid cell divisions may be influenced by its posttranslational modification status (Figure 1) and is certainly accomplished in coordination with other transcription factors.
Although separate roles for EKLF during the proliferation (activation of E2F2) versus differentiation (activation of p18 and p21) stages of erythroid differentiation may be operant, the role of cell-cycle components in erythroid differentiation is complex and does not follow the course typically observed in other cell types for renewal divisions (eg, E2F269 and E2F468 ). Indeed, the role of E2Fs may be heavily dependent on the cellular context in which they are expressed,74 effecting a dual functionality in progenitor cells compared with differentiating cells.75
EKLF in enucleation
EKLF function remains crucial for the final steps of erythroid maturation. Among several relevant genes affected by absence of EKLF is ICAM4,25,26,30 which codes an intercellular adhesion molecule that is critical for erythroblast/macrophage interactions within the erythroblastic island.76 Consistent with this requirement, peripheral blood from a congenital dyserythropoietic anemia (CDA) patient containing a missense EKLF mutation (described further in the next section) exhibited an abnormally high level of nucleated erythroblasts.77
EKLF mutations related to hematologic parameters and disease
Given the importance of EKLF during erythropoiesis, it is not surprising that several mutations within the mouse and human EKLF/KLF1 locus have recently been shown to cause hematologic changes and disorders (summarized in Table 1). These vary in their severity depending on the nature of the mutation. We have divided these mutations into 2 major categories based on the mechanisms that lead to the altered phenotypes, as shown in Figure 4.
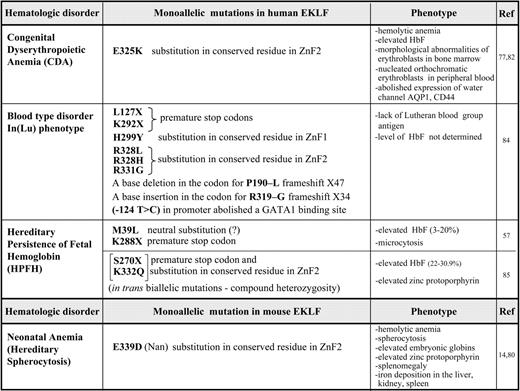

Mechanisms linking mutations in the EKLF/KLF1 gene with the impact that they have on erythrocyte phenotypes. The flow of gene expression from each of the 2 EKLF/KLF1 alleles at the top is shown as it progresses through transcript and protein expression, ultimately leading to the resultant phenotype and disorder as described in “EKLF mutations related to hematologic parameters and disease.” Selected examples from Table 1 are shown at the bottom that illustrate each mechanistic subtype. (A) A mutation (red) changes a functionally important amino acid (aa) and leads to an EKLF protein with altered properties that even in the presence of the WT allele causes a dominant mutant phenotype on a select subset of target genes. (B) Haploinsufficiency of EKLF follows as a consequence of: B(1), a mutation (green) that causes a premature stop codon in one allele, and the defective transcript undergoes nonsense mediated decay, leaving transcript from only a single WT allele to be translated into functional protein; B(2), a mutation (yellow) changes a structurally important amino acid and results in expression of a functionally impaired protein that decreases the pool of active EKLF to haploinsufficient levels. Positions of the mutations in the schematic are symbolic and do not relate to any precise amino acids.
Mechanisms linking mutations in the EKLF/KLF1 gene with the impact that they have on erythrocyte phenotypes. The flow of gene expression from each of the 2 EKLF/KLF1 alleles at the top is shown as it progresses through transcript and protein expression, ultimately leading to the resultant phenotype and disorder as described in “EKLF mutations related to hematologic parameters and disease.” Selected examples from Table 1 are shown at the bottom that illustrate each mechanistic subtype. (A) A mutation (red) changes a functionally important amino acid (aa) and leads to an EKLF protein with altered properties that even in the presence of the WT allele causes a dominant mutant phenotype on a select subset of target genes. (B) Haploinsufficiency of EKLF follows as a consequence of: B(1), a mutation (green) that causes a premature stop codon in one allele, and the defective transcript undergoes nonsense mediated decay, leaving transcript from only a single WT allele to be translated into functional protein; B(2), a mutation (yellow) changes a structurally important amino acid and results in expression of a functionally impaired protein that decreases the pool of active EKLF to haploinsufficient levels. Positions of the mutations in the schematic are symbolic and do not relate to any precise amino acids.
Dominant phenotype
The most extreme phenotype is generated by mutations in which a functionally important amino acid is replaced, leading to a protein with altered activation properties (Figure 4A). Two recent examples vividly illustrate this point. First, the mouse severe neonatal anemia (Nan) mutant exhibits a hemolytic anemia that displays many of the features of hereditary spherocytosis.14,78 Defects in structural membrane skeleton genes provide the molecular explanation for the associated red blood cell fragility.79 As a result, it came as a surprise to find not only that the primary defect in the Nan mouse maps to the EKLF gene, but also that it presents as a dominant phenotype despite the presence of the WT EKLF allele.14,80 Second, CDA is an inherited red blood cell disorder with the hallmarks of morphologic abnormalities of erythroblasts in the bone marrow and ineffective erythropoiesis and hemolysis, which can be divided into 3 major categories based on the erythroblast nuclear morphology.81 The genetic explanation for the more prevalent CDA1 and 2 categories have been determined, but analysis of unclassified CDA patients enabled their causative mutation to be mapped to EKLF,77,82 again displaying a dominant phenotype even in the presence of the WT EKLF allele. The remarkable aspect of these studies is that the mutations lead to a change in the same amino acid: E339 in mouse EKLF is equivalent to E325 in human EKLF. Glutamic acid at this site is absolutely conserved across EKLF proteins from different species and across the entire KLF family. This amino acid is located within the second zinc finger of mouse EKLF, precisely within the structural α-helical motif that fits into the major groove, enabling a direct interaction with the middle nucleotide of the EKLF-binding motif.12 However, the amino acid substitution is not the same in CDA patients as in Nan-EKLF and is of the opposite charge (aspartic acid in the mouse, lysine in the human), leading to intrinsic differences in DNA recognition and in the repertoire of affected target genes and resultant pathology.
Effects of the mouse E339D mutation have been characterized by biochemical tests and structural modeling of EKLF.14 Although conservative, this change alters the DNA-binding specificity of Nan-EKLF, limiting transcription to a select subset of EKLF target genes based on a subtle change in its cognate DNA element, in which a single nucleotide change marks the difference between recognition by both WT and Nan-EKLF (at 5′NGG GCG NGG) versus recognition by WT EKLF alone (at 5′NGG GTG NGG).14 An unusual observation is that even when expressed at an equivalent level to WT EKLF, Nan-EKLF selectively interferes with the expression of target genes with cognate elements that are not recognized by the mutant. However, because some target genes remain unaffected by Nan-EKLF, this mutant cannot act as a dominant-negative. The inability of the single WT copy to maintain normal expression at some but not all target sites in the Nan mouse also differentiates this mechanism from haploinsufficiency (described in the next section), particularly given the profound effects of Nan-EKLF on select targets that are, at most, mildly sensitive to EKLF dosage. Among the target genes most affected are E2f2, BKLF/Klf3, and the red cell membrane protein dematin, the low expression level of which partly explains the spherocytosis and hemolysis seen in these mice.14 There is a high level of residual embryonic globin expression in Nan. The net result is a distorted genetic output, anemia, and altered hemoglobin switching.14
A different pathology is exhibited by the human E325K mutation, which leads to abolished expression of the water channel AQP1 and of the adhesion molecule CD44 and reduced expression of 2 other adhesion molecules, BCAM and ICAM4. The peripheral blood contains evidence of poikilocytosis, anisocytosis, fragmented erythrocytes, and a large number of nucleated red blood cells, mostly orthochromatic erythroblasts, which suggests a failure of terminal erythroid differentiation. Nevertheless, there is some similarity to the Nan mouse; for example, β-like globin expression is dysregulated and CDA patients exhibit high expression levels of fetal and embryonic hemoglobins.77,82
Haploinsufficiency
Another category of EKLF mutation is more directly related to haploinsufficiency, and follows from 2 generative mechanisms (Figure 4B). Historically, EKLF WT and heterozygote (+/−) embryos and adults were considered indistinguishable (although hints of dose-dependent effects had been noted83 ), but recent studies conclusively show that both primitive30 and definitive erythropoiesis are sensitive to EKLF heterozygosity, primarily because the expression of a select set of EKLF target genes is sensitive to the reduced cellular levels of EKLF. For example, the rare blood group In(Lu) phenotype is caused by inheritance of a loss-of-function mutation on one allele of EKLF. Nine different mutations are found in the EKLF locus region. Some of them contain premature stop codons or frameshift mutations that lead to nonsense mediated decay of the transcript, leaving a single normal allele that results in a reduced level of EKLF protein (category B1 in Figure 4B). The presence of a mutated EKLF allele is easily detected by the almost complete absence of the Lutheran blood group antigen on the red cells. Other genes are significantly but not as dramatically affected, such as Alas2, AHSP, GPA, and Band3. Beyond this, the In(Lu) phenotype appears to be benign, indicating that one functional EKLF allele is sufficient to sustain human erythropoiesis.84
As mentioned previously, a subset of HPFH in a Maltese family is associated with K288X, a nonsense mutation in the EKLF gene that ablates its DNA-binding domain. Degradation of the transcript by nonsense-mediated decay leads to a haploinsufficient output (category B1). Expression profiling on primary erythroid progenitors shows that EKLF target genes are down-regulated in samples from individuals with HPFH, among them the BCL11A gene that encodes a suppressor of HbF expression. These observations provide an explanation for the effects of EKLF haploinsufficiency on elevated fetal hemoglobin levels.57
A subset of EKLF missense mutations leading to the In(Lu) phenotype yield an EKLF protein that is structurally compromised (category B2, Figure 4B). For example, the H299Y mutation is located at a critical amino acid required for zinc coordination and integrity of the finger structure.84 Practically speaking, this also generates a haploinsufficient condition, and dose-dependent target genes (such as the Lutheran antigen) are the most dramatically affected.
An interesting mixture of the 2 subsets of category B is found in a Sardinian family. High levels of fetal hemoglobin and a high level of zinc protoporphyrin are present only in compound hererozygotes that contain 2 EKLF mutations in trans: a S270X nonsense (category B1) and a K332Q missense (category B2) mutation. The K332Q mutation alters the efficiency of EKLF binding to the BCL11A, but not the adult β-globin CAC element. Surprisingly, monoallelic, isolated mutations of S270X or K332Q are associated with normal (low) fetal hemoglobin levels.85 These observations contradict the Maltese family data and suggest that genetic modifiers may play a role in the penetrance and expressivity of EKLF heterozygosity. For example, the HBS1L-MYB locus contains SNPs associated with HPFH.86 Because EKLF is a target of c-myb transcriptional activation,87 one possible scenario is that individuals harboring altered c-myb levels could modify the downstream effects of EKLF by subtle influence on its own critical concentration.
It is not known what percentage of HPFH variability is accounted for by EKLF mutation. However, scanning the human genome browser SNP database (dbSNP132) failed to reveal the Table 1 variants, indicating that they are not present in more than 1200 alleles analyzed so far in the 1000 genomes project. The accessibility of unpublished EKLF gene variants deposited into the HbVar Database88 will expand this list and may lead to testable hypotheses related to EKLF structure/function at the β-like globin locus.
The global EKLF network suggests other ways in which EKLF may behave as a genetic modifier. For example, variable levels of AHSP play a quantitative role in the severity of β-thalassemia intermedia, particularly in patients with triplicated α-globin genes.89 Because AHSP is strongly regulated by EKLF, variations in EKLF levels might attenuate or accentuate the protective effect of AHSP.
Conclusion
EKLF/KLF1 plays an essential role in coordinating the intracellular events that occur during erythropoiesis,90 particularly in directing megakaryocyte/erythroid progenitor lineage decisions, in establishing proper levels of morphologic and metabolic proteins, and in its continued requirement during the renewal and differentiative steps of terminal maturation. Coupled with the altered mammalian hematology that follows from mutated or haploinsufficient levels of EKLF/KLF1, these studies suggest that EKLF/KLF1 deficiency may be causative for a range of hematologic disorders.
Acknowledgments
The authors are grateful to Deepa Manwani, Jane Little, Shefali Soni, and Nithya Gnanapragasam for critical reading of this manuscript.
Referenced studies in the laboratory were supported by National Institutes of Health grants DK46865, DK48721, and DK77822 (to J.J.B.).
National Institutes of Health
Authorship
Contribution: M.S. and J.J.B. cowrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: James J. Bieker, Department of Developmental and Regenerative Biology, Mount Sinai School of Medicine, One Gustave Levy Pl, Box 1020, New York, NY 10029; e-mail: james.bieker@mssm.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal