

Abstract
We investigated age-related EBV+ B-cell lymphoproliferations in the Western population. The clinical features, histology, immunophenotype, EBV-encoded RNA in situ hybridization, and clonality by PCR of T-cell receptor gamma and immunoglobulin genes were categorized in 122 EBV+ lesions as follows: (1) reactive lymphoid hyperplasia; (2) polymorphic extranodal or (3) polymorphic nodal lymphoproliferative disease (LPD); and (4) diffuse large B-cell lymphoma (DLBCL). Interphase FISH for IG and PAX5 gene rearrangements was performed on 17 cases of DLBCL. The overall median age was 75 years (range, 45-101 years; 67 men, 55 women), and 67, 79, 73, and 77 years, respectively, for groups 1 through 4. Sixteen of 21 cases of polymorphic extranodal LPD were classified as EBV+ mucocutaneous ulcer. PCR for immunoglobulin genes was polyclonal in reactive lymphoid hyperplasia (84%) and monoclonal in 33%, 63%, and 56% of polymorphic extranodal and nodal LPD cases and DLBCL, respectively. All groups showed restricted/clonal T-cell receptor responses (27%-70%). By FISH, 19% of DLBCLs showed IGH@ rearrangements, but PAX5 was unaffected. Disease-specific 5-year survival was 100%, 93%, 57%, and 25% for groups 1-4, respectively, and 100% for patients with EBV+ mucocutaneous ulcer. Disease volume was predictive of therapy response (P = .0002), and pathologic subtype was predictive of overall outcome (P = .001). Age-related EBV+ B-cell LPD encompasses a wider disease spectrum than previously recognized and includes both reactive and neoplastic conditions. Reduction in the T-cell repertoire may contribute to decreased immune surveillance.
Introduction
After primary infection at an early age, EBV persists in a small proportion of B cells. The virus may elicit both T- and B-lymphocyte transformation and proliferation through complex mechanisms.1-4 A spectrum of EBV-driven B-cell lymphoproliferative disorders (LPDs) occurs in immunosuppressed patients with primary immune deficiency, HIV infection, or iatrogenic posttransplantation immunosuppression or who have received other treatments including methotrexate and tumor necrosis factor-α antagonists.5 Oyama et al recently described EBV+ B-cell LPDs in elderly Japanese patients,6 now recognized as a provisional entity in the World Health Organization classification: EBV+ diffuse large B-cell lymphoma (DLBCL) of the elderly.5 This is defined as a histologically malignant polymorphic or monomorphic B-cell lymphoproliferation in patients older than 50 years of age without any known immunodeficiency or prior lymphoma. The clinical behavior was aggressive, with frequent extranodal presentation and overall poor prognosis. There was some morphologic overlap with classic Hodgkin lymphoma (cHL), also encountered in the elderly but reported to have a better prognosis.7 The underlying immunologic deficit in this setting is believed to be immunosenescence, the natural decay of the immune system as a consequence of aging.2,6
We describe 122 cases of EBV+ B-cell LPDs in adults with no other identifiable cause of immunosuppression. Our aims were to analyze the demographic, clinical, and pathologic features not previously described in the Western population. We report that age-related EBV+ B-cell lymphoproliferative disorders (AR-EBVLPDs) represents a wider clinicopathologic spectrum than previously appreciated. They include reactive LPDs, as well as LPDs in some distinctive extranodal sites with differing prognoses. Comparisons with EBV+ B-cell LPDs in other settings of immunosuppression and the differential diagnoses with cHL and other LPDs are addressed, including issues related to pathogenesis, prognosis, and management.
Methods
Case selection
EBV-associated lymphoid proliferations in patients more than 45 years of age without evidence of acute EBV infection were selected from the files of the Laboratory of Pathology of the National Cancer Institute (Bethesda, MD), nearly all of which were submitted for diagnostic consultation over a 15-year period (1994-2009). The study was approved by the Institutional Review Board of the National Cancer Institute, South East Wales Research Ethics Committee, and the Cardiff and Vale University Health Board Research and Development Office. The cases originated from the United States, except for 1 case each from Italy, Spain, Singapore, South Korea, and Chile. Three cases were added from the files of the All Wales Lymphoma Panel (Cardiff, United Kingdom). The initial selection of B-cell LPDs was obtained by an electronic search of pathology reports for “EBV,” “EBER,” or “LMP1.” During the study period, EBV testing had been performed routinely for atypical lymphoid hyperplasias; atypical cutaneous, mucosal, or extranodal lesions; and in a proportion of B-cell lymphomas. The following categories were excluded: patients with a previous history of lymphoma; lymphoma entities in which EBV positivity represents a diagnostic requirement (lymphomatoid granulomatosis; DLBCL associated with chronic inflammation; primary effusion lymphoma; HIV-associated plasmablastic lymphoma; cHL); and cases with evidence of acute or recent EBV infection.
Histologic review was conducted by 2 of the authors (S.D.D. and E.S.J.), and lesions were classified into 4 diagnostic categories: (1) EBV-associated reactive lymphoid hyperplasia (RH), or lymph nodes with retained architecture in which the number of EBV-encoded RNA (EBER)–positive lymphoid cells exceeded that seen in normal lymph nodes from nonimmunosuppressed patients8,9 ; (2) EBV-associated extranodal polymorphic lymphoproliferative disease (Poly-E) well-circumscribed, isolated, and superficial ulcerating lesions that presented in the skin or mucosal surfaces were classified as EBV-associated mucocutaneous ulcer [EBVMCU], as defined previously10 ); (3) EBV-associated nodal polymorphic lymphoproliferative disease (Poly-N)6,11 ; and (4) EBV+ DLBCL.5 Patients were treated in their home institution by use of standard-of-care approaches.
Immunohistochemistry and in situ hybridization
Immunostaining on paraffin sections was performed for CD3, CD4, CD8, CD20, CD79a, MUM1, PAX5, Oct-2, Bob.1, CD138, EBV-LMP1, CD30, CD15, Ki67, κ, λ, CD10, Bcl-6, and Bcl-2 with an automated immunostainer (Ventana Medical Systems), with antigen retrieval and antibody dilutions performed per the manufacturer's recommendations and as published elsewhere.12 In situ hybridization for EBER was conducted on formalin-fixed, paraffin-embedded sections with an FITC-labeled oligonucleotide probe supplied by Ventana on an automated stainer (Ventana Benchmark). Visualization was achieved with the ISH iView system with alkaline phosphatase and NBT/BCIP substrate, with Fast Red as contrast.10
Quantification of EBER expression was assessed by computerized morphometric analysis of digital images. Three representative microscopic fields from each case were photographed with a 20× objective lens that covered a photographic area of 0.57 mm2. The digital images were “thresholded” to select the positive nuclei by use of ImageJ software Version 1.41o (National Institutes of Health; http://rsb.info.nih.gov/ij). An automated count of the selected nuclei was obtained, expressed as an average per millimeter squared. EBER positivity scored as above normal was > 10 EBER+ nuclei per 0.5 cm2 (> 0.2/mm2).8,9
PCR for immunoglobulin and TRG@ gene rearrangements
DNA was extracted from formalin-fixed, paraffin-embedded tissue blocks and amplified by PCR for detection of immunoglobulin (IGH@ and IGK@) and T-cell receptor-γ gene (TRG@) rearrangements, as published previously.11 The results were interpreted as polyclonal, restricted, or clonal. The “restricted” TRG@ category was defined as an abnormal rearrangement pattern with 1 or 2 small peaks that did not meet criteria for monoclonality, or an oligoclonal (multiple peaks) pattern.
Fluorescent in situ hybridization
FISH on formalin-fixed, paraffin-embedded sections followed standard protocols.13,14 The applied dual-color break-apart assays included LSI IGH@ (14q32 region; Abbott Molecular) and the previously described in-house assays for IGK@ (2p11), IGL@ (22q11),13 and PAX5 (9p13).15 Noncommercial probes were labeled directly with Spectrum Orange or Spectrum Green d-UTP (Abbott Molecular) by random priming. FISH images were acquired with a fluorescence microscope (Carl Zeiss) equipped with an Axiophot 2 camera (Carl Zeiss) and an Isis imaging system (MetaSystems).
Statistical analysis
The demographic features (Table 1), clinical parameters, and outcomes were analyzed with SAS version 9.1.3 software. The χ2 and Kruskal-Wallis tests were used for categorical and continuous variables, respectively. Unstratified univariate comparisons of survival between the pathologic groups were conducted with the log-rank test, with survival data displayed with Kaplan-Meier curves. Multivariate analyses that adjusted for other prognostic features were performed with Cox regression, with pathologic subgroup added last to the model. P < .05 was considered significant. Comparisons were made against DLBCL as the largest subgroup in the study.
Demographic features, presentation, management, clinical course, and outcome
| RH | Poly-E | Poly-N | DLBCL | ALL | P for heterogeneity* | ||
|---|---|---|---|---|---|---|---|
| Poly-E with EBVMCU | EBVMCU | ||||||
| Demographics | |||||||
| N | 31 | 21 | 16 | 30 | 40 | 122 | |
| Age (range), y | 67 (45-90) | 79 (58-101) | 79 (64-101) | 73 (48-93) | 77 (59-90) | 75 (45-101) | .12† |
| Male/female, n | 16/15 | 9/12 | 6/10 | 13/17 | 29/11 | 67/55 | .046‡ |
| Involved site, n | 31 | 21 | 16 | 30 | 40 | 122 | |
| Extranodal, n (%) | 0 (0) | 21 (100) | 16 (100) | 0 (0) | 14 (35) | 35 (29) | |
| Nodal, n (%) | 31 (100) | 0 (0) | 0 (0) | 30 (100) | 24 (60) | 85 (69) | |
| Nodal and extranodal, n (%) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 2 (5) | 2 (2) | < .0001, any nodal involvement‡ |
| Extent of disease, n | 31 | 21 | 16 | 23 | 33 | 108 | |
| Localized, n (%) | 20 (65) | 16 (76) | 14 (87) | 10 (44) | 12 (37) | 58 (54) | .01, local vs not† |
| Tongue, n | 0 | 4 | 4 | 0 | 0 | 0 | |
| Tonsil, n | 0 | 4 | 4 | 0 | 0 | 0 | |
| Oral/palate/nasopharynx, n | 0 | 3 | 1 | 0 | 0 | 0 | |
| Skin, n | 0 | 5 | 5 | 0 | 0 | 0 | |
| Bulky localized, n (%) | 1 (3) | 1 (5) | 0 (0) | 0 (0) | 3 (9) | 5 (5) | |
| Two sites, n (%) | 1 (3) | 2 (9) | 2 (13) | 0 (0) | 3 (9) | 6 (5) | |
| Multiple sites, n (%) | 1 (3) | 1 (5) | 0 (0) | 4 (17) | 1 (3) | 7 (6) | |
| Generalized, n (%) | 8 (26) | 1 (5) | 0 (0) | 9 (39) | 14 (42) | 32 (30) | |
| B symptoms, n (%) | 10 (32) | 0 (0) | 0 (0) | 8 (35) | 12 (36) | 30 (28) | .02‡ |
| Management, n | 24 | 18 | 14 | 16 | 20 | 78 | |
| No treatment, n (%) | 19 (79) | 10 (55) | 9 (64) | 6 (38) | 0 (0) | 35 (45) | < .0001, treated vs not‡ |
| Chemotherapy, n (%) | 5 (21) | 2 (11) | 1 (7) | 9 (56) | 16 (80) | 32 (41) | |
| Radiotherapy, n (%) | 0 (0) | 4 (22) | 3 (21) | 0 (0) | 1 (5) | 5 (6) | |
| Chemotherapy + radiotherapy, n (%) | 0 (0) | 1 (6) | 1 (7) | 1 (6) | 2 (10) | 4 (5) | |
| Bone marrow transplantation, n (%) | 0 (0) | 1 (6) | 0 (0) | 0 (0) | 1 (5) | 2 (3) | |
| Course, n | 21 | 18 | 14 | 16 | 20 | 75 | |
| Spontaneous resolution, n (%) | 14 (67) | 6 (28) | 5 (36) | 3 (20) | 0 (0) | 23 (30) | |
| Relapsing/remitting, n (%) | 0 (0) | 3 (17) | 3 (21) | 0 (0) | 0 (0) | 3 (4) | |
| Complete remission [% of treated], n (%) | 4 (19) [100] | 6 (38) [75] | 5 (36) [100] | 6 (38) [60] | 11 (55) [55] | 27 (36) [63] | < .0001‡ |
| Partial remission [% of treated], n (%) | 0 (0) [0] | 1 (5) [13] | 0 (0) [0] | 0 (0) [0] | 4 (20) [20] | 5 (7) [12] | |
| Stable persistent disease, n (%) | 3 (14) | 1 (5) | 1 (7) | 1 (6) | 0 (0) | 5 (7) | |
| Progressive disease, n (%) | 0 (0) | 1 (5) | 0 (0) | 6 (38) | 5 (25) | 12 (16) | |
| Histologic transformation, n (%) | 3 (9) | 0 (0) | 0 (0) | 3 (10) | 0 (0) | 6 (5) | .56‡ |
| to Poly-N, n | 1 | 0 | 0 | 0 | 0 | 1 | |
| to DLBCL, n | 0 | 0 | 0 | 3 | 0 | 3 | |
| to cHL, n | 2 | 0 | 0 | 0 | 0 | 2 | |
| Outcomes, n | 17 | 15 | 14 | 13 | 18 | 63 | |
| Died of disease, n (%) | 0 (0) | 1 (7) | 0 (0) | 4 (31) | 7 (39) | 12 (19 | |
| Died of complications of Tx, n (%) | 0 (0) | 0 (0) | 0 (0) | 1 (8) | 2 (11) | 3 (5) | |
| Died of unrelated cause, n (%) | 1 (6) | 6 (40) | 6 (43) | 3 (23) | 1 (6) | 11 (17) | |
| Median (range) follow-up, mo | 36 (2-132) | 38 (2-72) | 38 (2-60) | 71 (40-101) | 24 (1-68) | 41 (1-132) | .2§ |
| Median survival, mo | NR | 24 | 21 | 24 | 25 | 53 | .009§ |
| RH | Poly-E | Poly-N | DLBCL | ALL | P for heterogeneity* | ||
|---|---|---|---|---|---|---|---|
| Poly-E with EBVMCU | EBVMCU | ||||||
| Demographics | |||||||
| N | 31 | 21 | 16 | 30 | 40 | 122 | |
| Age (range), y | 67 (45-90) | 79 (58-101) | 79 (64-101) | 73 (48-93) | 77 (59-90) | 75 (45-101) | .12† |
| Male/female, n | 16/15 | 9/12 | 6/10 | 13/17 | 29/11 | 67/55 | .046‡ |
| Involved site, n | 31 | 21 | 16 | 30 | 40 | 122 | |
| Extranodal, n (%) | 0 (0) | 21 (100) | 16 (100) | 0 (0) | 14 (35) | 35 (29) | |
| Nodal, n (%) | 31 (100) | 0 (0) | 0 (0) | 30 (100) | 24 (60) | 85 (69) | |
| Nodal and extranodal, n (%) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 2 (5) | 2 (2) | < .0001, any nodal involvement‡ |
| Extent of disease, n | 31 | 21 | 16 | 23 | 33 | 108 | |
| Localized, n (%) | 20 (65) | 16 (76) | 14 (87) | 10 (44) | 12 (37) | 58 (54) | .01, local vs not† |
| Tongue, n | 0 | 4 | 4 | 0 | 0 | 0 | |
| Tonsil, n | 0 | 4 | 4 | 0 | 0 | 0 | |
| Oral/palate/nasopharynx, n | 0 | 3 | 1 | 0 | 0 | 0 | |
| Skin, n | 0 | 5 | 5 | 0 | 0 | 0 | |
| Bulky localized, n (%) | 1 (3) | 1 (5) | 0 (0) | 0 (0) | 3 (9) | 5 (5) | |
| Two sites, n (%) | 1 (3) | 2 (9) | 2 (13) | 0 (0) | 3 (9) | 6 (5) | |
| Multiple sites, n (%) | 1 (3) | 1 (5) | 0 (0) | 4 (17) | 1 (3) | 7 (6) | |
| Generalized, n (%) | 8 (26) | 1 (5) | 0 (0) | 9 (39) | 14 (42) | 32 (30) | |
| B symptoms, n (%) | 10 (32) | 0 (0) | 0 (0) | 8 (35) | 12 (36) | 30 (28) | .02‡ |
| Management, n | 24 | 18 | 14 | 16 | 20 | 78 | |
| No treatment, n (%) | 19 (79) | 10 (55) | 9 (64) | 6 (38) | 0 (0) | 35 (45) | < .0001, treated vs not‡ |
| Chemotherapy, n (%) | 5 (21) | 2 (11) | 1 (7) | 9 (56) | 16 (80) | 32 (41) | |
| Radiotherapy, n (%) | 0 (0) | 4 (22) | 3 (21) | 0 (0) | 1 (5) | 5 (6) | |
| Chemotherapy + radiotherapy, n (%) | 0 (0) | 1 (6) | 1 (7) | 1 (6) | 2 (10) | 4 (5) | |
| Bone marrow transplantation, n (%) | 0 (0) | 1 (6) | 0 (0) | 0 (0) | 1 (5) | 2 (3) | |
| Course, n | 21 | 18 | 14 | 16 | 20 | 75 | |
| Spontaneous resolution, n (%) | 14 (67) | 6 (28) | 5 (36) | 3 (20) | 0 (0) | 23 (30) | |
| Relapsing/remitting, n (%) | 0 (0) | 3 (17) | 3 (21) | 0 (0) | 0 (0) | 3 (4) | |
| Complete remission [% of treated], n (%) | 4 (19) [100] | 6 (38) [75] | 5 (36) [100] | 6 (38) [60] | 11 (55) [55] | 27 (36) [63] | < .0001‡ |
| Partial remission [% of treated], n (%) | 0 (0) [0] | 1 (5) [13] | 0 (0) [0] | 0 (0) [0] | 4 (20) [20] | 5 (7) [12] | |
| Stable persistent disease, n (%) | 3 (14) | 1 (5) | 1 (7) | 1 (6) | 0 (0) | 5 (7) | |
| Progressive disease, n (%) | 0 (0) | 1 (5) | 0 (0) | 6 (38) | 5 (25) | 12 (16) | |
| Histologic transformation, n (%) | 3 (9) | 0 (0) | 0 (0) | 3 (10) | 0 (0) | 6 (5) | .56‡ |
| to Poly-N, n | 1 | 0 | 0 | 0 | 0 | 1 | |
| to DLBCL, n | 0 | 0 | 0 | 3 | 0 | 3 | |
| to cHL, n | 2 | 0 | 0 | 0 | 0 | 2 | |
| Outcomes, n | 17 | 15 | 14 | 13 | 18 | 63 | |
| Died of disease, n (%) | 0 (0) | 1 (7) | 0 (0) | 4 (31) | 7 (39) | 12 (19 | |
| Died of complications of Tx, n (%) | 0 (0) | 0 (0) | 0 (0) | 1 (8) | 2 (11) | 3 (5) | |
| Died of unrelated cause, n (%) | 1 (6) | 6 (40) | 6 (43) | 3 (23) | 1 (6) | 11 (17) | |
| Median (range) follow-up, mo | 36 (2-132) | 38 (2-72) | 38 (2-60) | 71 (40-101) | 24 (1-68) | 41 (1-132) | .2§ |
| Median survival, mo | NR | 24 | 21 | 24 | 25 | 53 | .009§ |
ALL indicates acute lymphoblastic leukemia; Tx, treatment; and NR, (median) not reached.
Comparing RH vs Poly-E vs Poly-N vs DLBCL.
Kruskal-Wallis test.
χ2 test.
Unadjusted log-rank test.
Results
Histologic, immunophenotypic, and genotypic features
EBV-associated RH (31 cases).
Lymph nodes showed preserved architecture with a range of reactive patterns that included follicular hyperplasia (45%), paracortical hyperplasia (43%), regressive follicular changes resembling Castleman disease (6%), and plasma cell hyperplasia (6%; Figure 1A-C). Monocytoid B-cell reaction and epithelioid granulomas were variably present. The paracortical infiltrate was polymorphous, with frequent immunoblasts and occasional Hodgkin-Reed Sternberg (HRS)–like cells. Immunostains highlighted intact architecture. CD20 outlined prominent interfollicular immunoblasts, frequently positive for CD30 but without expression of CD15 (Table 2). Staining for EBER was either restricted to focal germinal centers (26% of cases) or scattered through the paracortex (Figure 2G-H). Polyclonal plasma cells were present but EBER−. The average EBV count was 34.3/mm2 (Table 2). The majority of the cases tested were polyclonal for both IG and TRG@ by PCR (84% and 73%, respectively). Twenty-seven percent of cases had evidence of a clonal T-cell population (Figure 3).

Histologic features of AR-EBVLPD. (A) Reactive follicular hyperplasia. (B) Reactive paracortical hyperplasia. (C) Plasma cell hyperplasia: monomorphic proliferation of uniform plasma cells. (D) Poly-N: polymorphous infiltrate comprising plasma cells, lymphocytes, and immunoblasts with occasional eosinophils. Occasional atypical HRS-like cells are noted (inset). (E) Poly-E–EBVMCU: well-circumscribed ulcerated lesion in oral mucosa with polymorphous infiltrate and HRS-like cells (inset). (F) DLBCL: “conventional” histologic picture with a diffuse proliferation of large mononuclear cells with centroblastic and immunoblastic features. Occasional pleomorphic cells are noted, but an inflammatory background is absent. (G) DLBCL: large numbers of HRS-like cells are present. (H) Plasmablastic lymphoma: the cells show basophilic cytoplasm and most have prominent nuclei (H&E; original magnifications, ×20 (B,E); ×40 (A); and ×400 (C,D,F-H and insets).
Histologic features of AR-EBVLPD. (A) Reactive follicular hyperplasia. (B) Reactive paracortical hyperplasia. (C) Plasma cell hyperplasia: monomorphic proliferation of uniform plasma cells. (D) Poly-N: polymorphous infiltrate comprising plasma cells, lymphocytes, and immunoblasts with occasional eosinophils. Occasional atypical HRS-like cells are noted (inset). (E) Poly-E–EBVMCU: well-circumscribed ulcerated lesion in oral mucosa with polymorphous infiltrate and HRS-like cells (inset). (F) DLBCL: “conventional” histologic picture with a diffuse proliferation of large mononuclear cells with centroblastic and immunoblastic features. Occasional pleomorphic cells are noted, but an inflammatory background is absent. (G) DLBCL: large numbers of HRS-like cells are present. (H) Plasmablastic lymphoma: the cells show basophilic cytoplasm and most have prominent nuclei (H&E; original magnifications, ×20 (B,E); ×40 (A); and ×400 (C,D,F-H and insets).
EBER count by histologic subtype
| Subtype | Mean | Median | Range |
|---|---|---|---|
| RH | 34.3 | 17 | 0.43-247.4 |
| Poly-E | 92.8 | 81 | 0.85-278.9 |
| Poly-N | 168.8 | 121 | 2.3-676.0 |
| DLBCL | 176.8 | 150 | 7.3-462.8 |
| Subtype | Mean | Median | Range |
|---|---|---|---|
| RH | 34.3 | 17 | 0.43-247.4 |
| Poly-E | 92.8 | 81 | 0.85-278.9 |
| Poly-N | 168.8 | 121 | 2.3-676.0 |
| DLBCL | 176.8 | 150 | 7.3-462.8 |
EBER count is per millimeter squared.
P < .001 for heterogeneity.

Immunohistochemical features and EBER in situ hybridization of AR-EBVLPD. (A-F) Poly-N with prominent HRS-like cells. (A) Reduced expression of CD20. (B) The HRS-like cells are CD30+. There is nuclear expression of PAX5 (C), Oct.2 (D), and BOB1 (E). (F) Occasional cells are positive for CD15. (G) Reactive follicular hyperplasia. Germinal center shows intense EBER positivity. (H) Reactive paracortical hyperplasia. There is paracortical/interfollicular distribution of EBER+ cells. (I) EBVMCU. Note superficial distribution of EBER+ cells. (J) Poly-N. High concentration of EBER+ cells. (K) Poly-E. Arterial wall infiltrated by EBER+ B cells. Original magnifications, ×20 (I); ×40 (G-H); ×100 (J-K); ×400 (A-E); and ×600 (F).
Immunohistochemical features and EBER in situ hybridization of AR-EBVLPD. (A-F) Poly-N with prominent HRS-like cells. (A) Reduced expression of CD20. (B) The HRS-like cells are CD30+. There is nuclear expression of PAX5 (C), Oct.2 (D), and BOB1 (E). (F) Occasional cells are positive for CD15. (G) Reactive follicular hyperplasia. Germinal center shows intense EBER positivity. (H) Reactive paracortical hyperplasia. There is paracortical/interfollicular distribution of EBER+ cells. (I) EBVMCU. Note superficial distribution of EBER+ cells. (J) Poly-N. High concentration of EBER+ cells. (K) Poly-E. Arterial wall infiltrated by EBER+ B cells. Original magnifications, ×20 (I); ×40 (G-H); ×100 (J-K); ×400 (A-E); and ×600 (F).

PCR for IG and TRG@ gene rearrangements. (A) The majority of the cases of RH were polyclonal for IG gene rearrangements (84%); Poly-E, Poly-N, and DLBCL showed clonal IG gene rearrangements in 33%, 63%, and 56% of cases, respectively. (B) 27% of cases with RH had monoclonal TRG@ gene rearrangements and no restricted T-cell responses; Poly-E, Poly-N, and DLBCL showed clonal TRG@ gene rearrangements in 50%, 24%, and 15% of cases, respectively; Poly-E, Poly-N, and DLBCL showed restricted T-cell responses in 20%, 9%, and 15% of cases, respectively.
PCR for IG and TRG@ gene rearrangements. (A) The majority of the cases of RH were polyclonal for IG gene rearrangements (84%); Poly-E, Poly-N, and DLBCL showed clonal IG gene rearrangements in 33%, 63%, and 56% of cases, respectively. (B) 27% of cases with RH had monoclonal TRG@ gene rearrangements and no restricted T-cell responses; Poly-E, Poly-N, and DLBCL showed clonal TRG@ gene rearrangements in 50%, 24%, and 15% of cases, respectively; Poly-E, Poly-N, and DLBCL showed restricted T-cell responses in 20%, 9%, and 15% of cases, respectively.
EBV-associated Poly-E (21 cases).
Most cases in this group (n = 16) fulfilled criteria for EBVMCU and were characterized by localized, sharply circumscribed cutaneous or oropharyngeal ulcers. The pathologic features of EBVMCU have been described elsewhere10 and are illustrated in Figures 1E and 2I. The 5 remaining cases were characterized by an extensive polymorphic infiltrate accompanied by necrosis, reminiscent of polymorphic posttransplantation lymphoproliferative disorder (PTLD). Immunoblasts were usually prominent, and HRS-like cells were present in 2 cases. Marked angioinvasion was noted in 3 cases, accompanied by extensive necrosis (Figure 2K). In situ hybridization for EBER highlighted numerous cells (81/mm2) with a range in size and morphology (Figure 2J). Positivity for EBER colocalized with the expression of LMP1, CD20, and PAX5. IG and TRG@ gene rearrangements were successful in 12 and 10 cases, respectively, with a monoclonal IG rearrangement in 33% and a monoclonal TRG@ gene rearrangement in 50%; 20% had a restricted TRG@ pattern (Figure 3).
EBV-associated Poly-N (30 cases).
In contrast to RH, these cases showed effaced architecture by an infiltrate reminiscent of polymorphic PTLD (Figure 1D). Lymphoid follicles were generally absent (85% of cases) or inconspicuous. HRS-like cells were seen in 63%, with prominent cytologic atypia in 23% (Figure 1D inset). Thirteen percent showed angioinvasion, particularly within the perinodal tissue (Figure 2K).
On immunostaining, there was reduction of expression of CD20 in the atypical blasts in 19% of cases, with all the cells showing positivity for CD79a. The pleomorphic cells were positive for CD30, MUM-1, and PAX5. EBER counts were approximately 5 times higher than those observed in RH (median 121/mm2; Table 2).
IG and TRG@ PCRs were successful in 24 and 29 cases, respectively. A clonal IG rearrangement was seen in 63%. On PCR for TRG@, 24% had a clonal rearrangement, and 9% of cases showed a restricted pattern (Figure 3).
EBV+ DLBCL (40 cases).
In 33 cases (82.5%), the features were those of DLBCL of the elderly. Lymph nodes (60%) were completely effaced, often accompanied by necrosis (35%). Extranodal sites showed similar diffuse permeation. The infiltrate displayed marked pleomorphism, lacking the spectrum of plasmacytoid differentiation and range in cell size seen in Poly-N. Seventeen cases (51.5%) contained HRS-like cells in addition to diffuse areas populated by pleomorphic mononuclear blasts (Figure 1F-G). Angioinvasion was seen in 21%. In 39%, the large B cells were widely scattered, at least in part, resembling T-cell/histiocyte-rich B-cell lymphoma. Thirty percent showed morphology of conventional DLBCL. Reduced staining for CD20 was seen in 19%, with strong positivity for CD30 in 88%. The B-cell phenotype was preserved with coexpression of PAX5, Oct-2, and Bob.1. CD15 was focally expressed in 25% but lacked other features typical for cHL, with the B blasts exhibiting a broad morphologic spectrum. Seven cases (18%) were classified as plasmablastic lymphoma, comprising a uniform population of large transformed cells, as described previously (Figure 1H).16 EBER counts were markedly elevated (median, 150/mm2). A result for IG and TRG@ PCR was obtained in 18 and 13 cases, respectively. A B-cell clone was shown in 56%. Fifteen percent of cases showed a monoclonal pattern and another 15% a restricted T-cell pattern.
To determine whether IGH@, IGK@, and IGL@ loci known to mediate chromosomal translocations in B-cell non-Hodgkin lymphoma were affected in EBV+ DLBCL, we performed interphase FISH analysis with the respective break-apart probes on formalin-fixed, paraffin-embedded sections from 17 cases. In addition, we applied a break-apart assay for PAX5 shown to be involved in t(9;14)(p13;q32) in 1 case of EBV+ DLBCL.17 FISH experiments were successful in 16 of 17 cases. A split of IGH@ signals that heralded an IGH@/14q32 translocation was found in 3 cases. The hybridization pattern of the remaining probes indicated recurrent copy number changes of the analyzed loci but not their rearrangements (Figure 4).

Examples of FISH with LSI IGH and IGL break-apart probes performed on case 18 (A) and case 42 (B), respectively. Note a split of IGH signals indicative of t(14q32/IGH) in panel A and copy number changes of IGL in panel B.
Examples of FISH with LSI IGH and IGL break-apart probes performed on case 18 (A) and case 42 (B), respectively. Note a split of IGH signals indicative of t(14q32/IGH) in panel A and copy number changes of IGL in panel B.
Pathologic features of recurrent disease, transformation, and other subsequent lymphomas
Thirteen patients had multiple biopsies during the course of the disease for the assessment of relapse or clinical progression. Two were diagnosed with Poly-N, with no histologic change at recurrence; however, 1 had evidence of monoclonal IG rearrangement by PCR in the second biopsy, the first being negative. One patient with polyclonal RH had a recurrence with Poly-N, showing loss of architecture, evidence of clonal IG rearrangement, and an increase in the EBER count from 0.4/mm2 to 113/mm2 in the recurrence. Three additional patients showed evidence of progression from Poly-N to DLBCL. Two had monoclonal IG rearrangements by PCR in the initial biopsy, with the same clone shown at relapse. Two patients initially diagnosed as having RH with EBV+ germinal centers relapsed with EBV+ cHL of mixed cellularity subtype. Both patients tested were polyclonal for IG rearrangements by PCR. One patient with Poly-E subsequently developed subcutaneous panniculitis-like T-cell lymphoma, negative for EBV. One patient with Poly-E and features of EBVMCU had a biopsy of an isolated neck lymphadenopathy that showed follicular hyperplasia with EBER+ germinal centers. Two patients with DLBCL had identical pathologic features in multiple biopsies, with plasmablastic lymphoma and DLBCL-NOS, respectively.
Clinical features and course
The initial screening identified 191 EBV+ LPDs. On the basis of selection criteria, 69 cases were excluded, which left a study cohort of 122 patients (67 men and 55 women; median age 75 years [range, 45-101 years]). Full follow-up information was available for 63 patients (51%). The demographic features, presentation, management, clinical course, and outcomes of these patients are summarized in Table 1.
EBV+ RH.
There were 31 patients in this group (16 men, 15 women; median age, 67 years; range, 45-90 years). They presented with localized lymphadenopathy (65%), bulky disease (3%), and generalized lymphadenopathy (26%). One-third had B symptoms. The cases were referred because of a suspicious clinical history, atypical histologic features, or both. In the majority, the course was self-limited and required no treatment (79%); however, on clinical grounds, 21% received treatment that included prednisolone, rituximab, and R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisolone). All treated patients achieved complete remission (CR). In 2 patients, lymphadenopathy persisted, with no progression after follow-up of 24 and 136 months, respectively. Two patients subsequently developed cHL, 2 and 60 months after initial biopsy, 1 having had longstanding persistent lymphadenopathy. One patient progressed to Poly-N 36 months after initial presentation. There were no disease-associated deaths in this group. One patient died of an unrelated cause.
EBV-associated Poly-E.
There were 21 patients in this group (9 men, 12 women; median age, 79 years; range, 58-101 years). Sixteen had localized disease (76%) with features of EBVMCU. Details of the patients with EBVMCU were reported previously; median age was 79 years (range, 64-101 years).11 Two of the 7 patients with cutaneous disease had involvement of underlying soft tissue or local extension. The clinical presentation was characterized by slowly developing indurated ulcers, which in 9 cases involved oropharyngeal mucosa (tongue [4], tonsils [4], or palate [1] only). Five patients had cutaneous lesions of the lips, arms, or chest. Two patients with oropharyngeal lesions in addition had concomitant isolated unilateral neck lymphadenopathy, but none had evidence of systemic lymphadenopathy, hepatosplenomegaly, or bone marrow involvement. Of the 16 patients with EBVMCU, 3 had multiple episodes of oral mucosal ulceration with spontaneous resolution. Nine did not receive any therapy. Of those with available follow-up (n = 14), 1 patient was deemed too frail for any treatment. He had stable persistent disease for 5 months and died of unrelated causes. After biopsy, there was spontaneous regression of 4 lesions with no further evidence of disease after a median follow-up of 18 months (range, 3-36 months). Five patients with EBVMCU were treated actively. One received chemotherapy, 1 had radiochemotherapy, and 3 had radiotherapy only. All treated patients remained in CR after a median follow-up of 15 months (range, 3-60 months).
The remaining 5 patients with Poly-E presented with lesions in the parotid region, skin, supraglottic area, nasopharynx, liver, and tongue. Two patients had widespread disease, and 3 had bulky localized disease. One patient received no treatment and underwent spontaneous resolution. Three patients received chemotherapy, radiotherapy, and bone marrow transplantation, respectively; 1 achieved CR, 1 achieved partial remission, and 1 had progressive disease.
EBV-associated Poly-N.
There were 30 patients in this group (13 men, 17 women; median age, 73 years; range, 48-93 years). Fifty-six percent presented with generalized lymphadenopathy, and 35% had B symptoms. The majority of the patients (62%) were treated actively with chemotherapy alone (56%) or with the addition of radiotherapy (6%). The chemotherapy regimens included R-prednisolone, CHOP, R-CHOP, and R-EPOCH (rituximab, etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin) with maintenance rituximab. Lymphadenopathy resolved spontaneously with no treatment in 20% of cases. CR was achieved in 60% of treated cases, with relapse seen in 1 case 1 year after treatment. One patient had stable disease (6%). Clinical progression was noted in 6 patients (38%), 2 of whom did not receive any treatment because of poor performance status. Three patients developed DLBCL 2, 6, and 13 months after initial diagnosis, respectively. Four patients (31%) died of disease, 1 (8%) of complications of chemotherapy, having initially had a good therapeutic response. Three patients (23%) died of unrelated causes. The median survival of this group was 24 months.
EBV+ DLBCL.
There were 40 patients in this group (29 men, 11 women; median age, 77 years; range, 59-90 years). Sixty percent presented with disease in nodal sites. Extranodal presentations (35%) included oropharynx and palate, maxillary sinus, gastrointestinal tract, skin, adrenals, liver, and lung; 71% of patients with plasmablastic features had extranodal disease. Fifty-four percent had multiple sites of involvement, often with B symptoms (36%). Most patients received chemotherapy with R-CHOP (80%), which was combined with radiotherapy in an additional 10% of cases. Radiotherapy alone was given in 5% of cases, and 1 patient (5%) was treated with bone marrow transplantation. CR was achieved in 55% of cases, 20% achieved partial remission, and 25% had progressive disease. One patient developed cHL 12 months after initial diagnosis. Thirty-nine percent of these patients had disease-related deaths, and an additional 11% died as a result of complications of chemotherapy. Six percent of patients died of unrelated causes. The median survival of this group was 25 months.
Statistical analysis
There was no statistical difference in the ages of the groups. There was a significant difference in the sex distribution between groups, caused mainly by a higher rate of male patients in the DLBCL group (P = .046; Table 1). The EBER count between all the groups was significantly different (P < .001), with RH having the lowest median score and DLBCL the highest (Table 2). The groups differed significantly for IG rearrangements (P = .005) but not for TRG@ gene rearrangements (P = .19 for heterogeneity; P = .11 for polyclonal versus restricted or clonal; Figure 3).
The volume of disease at presentation (localized, bulky localized, 2 sites, multiple sites, or generalized) correlated highly with the response to treatment, the poorest outcome being associated with generalized disease (P = .0002 for correlation). This translated into a significant difference in disease-related mortality (hazard ratio adjusted for age between groups 1.51, 95% confidence interval 1.10-2.06, P = .01).
In unadjusted univariate analyses, pathologic subtype correlated significantly with overall survival (P = .009; Table 1; Figure 5). In adjusted Cox regression analysis with allowance for age and volume of disease and with censoring of deaths not related to disease, pathologic subgroup was predictive of outcome (P = .001 for heterogeneity; Table 3). In adjusted analyses, there were also highly significant differences between diagnostic groups in overall survival. In model building, the other tested variables, including sex, IG or TRG@ clonality, and EBER count, were not found to be prognostic.
Results of multivariate Cox regression analyses
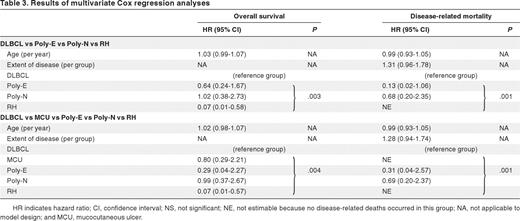
HR indicates hazard ratio; CI, confidence interval; NS, not significant; NE, not estimable because no disease-related deaths occurred in this group; NA, not applicable to model design; and MCU, mucocutaneous ulcer.

Kaplan-Meier survival curves. Curves are shown for (1) RH (Reactive), (2) Poly-E, (3) EBVMCU (MCU), (4) Poly-N, and (5) DLBCL. (A) Overall survival for groups 1, 2, 4, and 5. (B) Overall survival for groups 1-5. (C) Disease-related mortality for groups 1, 2, 4, and 5. (D) Disease-related mortality for groups 1-5. Plots for Reactive and MCU overlap in panel D.
Kaplan-Meier survival curves. Curves are shown for (1) RH (Reactive), (2) Poly-E, (3) EBVMCU (MCU), (4) Poly-N, and (5) DLBCL. (A) Overall survival for groups 1, 2, 4, and 5. (B) Overall survival for groups 1-5. (C) Disease-related mortality for groups 1, 2, 4, and 5. (D) Disease-related mortality for groups 1-5. Plots for Reactive and MCU overlap in panel D.
Discussion
Prior reports of AR-EBVLPD have come mainly from Japan, with only rare reports of this disease in Western populations.18 In the present series, the demographic and clinical features of Poly-N and DLBCL were largely in accordance with recent data from Oyama et al.6,11 The median ages (73 and 77 years) of the Poly-N and DLBCL patients in the present study were close to the median age of 71 years described by those authors6 and showed a similar preponderance of males (1.5:1.0 ratio of men to women) in the DLBCL group. There was a high prevalence of B symptoms (34%-37%) and multiple sites of involvement or generalized disease at presentation (74%-78%). A significant proportion of DLBCLs involved extranodal sites (40%). Poor response to therapy and poor overall survival were comparable for Poly-N and DLBCL patients.
The early report from Oyama et al11 found a much better prognosis for the group they identified as polymorphic than for those with EBV+ DLBCL; however, they failed to confirm the good prognosis of the polymorphic subtype in their subsequent series,6 a finding that was left unexplained. Our identification of a new subset of patients with localized extranodal disease who manifest features of EBVMCU and have a good prognosis appears to resolve this discrepancy. Although the entire group of Poly-E patients showed overall poor survival, patients who presented with localized ulcers in the skin and oropharyngeal mucosa stood out, with distinctive pathologic and clinical features including waxing and waning disease, high rate of spontaneous remission, and no disease-associated deaths. Interestingly, in the 2003 report from Oyama et al,11 at least 5 of 13 polymorphic patients presented with disease that involved skin and oropharynx, which possibly conformed to EBVMCU.
EBVMCU is a distinctive lesion that is part of the spectrum of AR-EBVLPD, and its typically self-limited clinical course warrants special attention. The hallmarks of EBVMCU are a sharply circumscribed and superficial ulcer that contains numerous HRS-like cells, often positive for both CD30 and CD15, accompanied by prominent apoptosis. A prominent rim of reactive T lymphocytes lies at the base. The cases in the present series showed a relatively low level of clonal IG rearrangements and a self-limited course; however, we cannot rule out a false-negative PCR result given the marked cytologic atypia observed in the EBV+ blasts. EBVMCU also occurs in other settings associated with iatrogenic immunosuppression (by methotrexate, cyclosporine A, and azathioprine), and in those patients in whom immunosuppression could be withdrawn, spontaneous resolution was observed.10
We also have made several other new observations. We identified a substantial number of patients with EBV-associated RH, a subset of patients with a good prognosis, who appear to be at low risk for progression to lymphoma. Patients with RH were 6-12 years younger than those in the other groups, mainly presented with localized lymphadenopathy (71%), exhibited a high rate of spontaneous resolution (67%), and had excellent overall survival. Early reactive lesions had been postulated on the basis of a small number of cases of lymphoid hyperplasia in the Japanese population, in whom the changes were interpreted to simulate autoimmune conditions.19 One patient with RH progressed to Poly-N, with the emergence of a clonal B-cell population in the subsequent lymph node biopsy. Interestingly, 2 patients with RH subsequently developed EBV+ cHL of the mixed-cellularity subtype. Expansion of EBV+ B cells in the RH lymph nodes may have portended the evolution to cHL, a sequence that has been proposed in acute infectious mononucleosis.20 The complex molecular mechanisms of EBV involvement in the pathogenesis of cHL have been widely studied.21
The postulated pathogenetic mechanism behind AR-EBVLPD is immunosenescence, a complex spectrum of regressive changes that affect the immune system with aging.22-24 T-cell responses appear to be the most profoundly affected, with the accumulation of clonal CD8+ T cells with mature memory cell phenotypes but diminished functionality. This occurs because of a reduced capacity to maintain the pool of new, naive T cells, which is compensated for by proliferation within the mature and senescent memory cell pool. The result is a prevalence of oligoclonal T cells (“restricted” T-cell response) that are markedly deficient in their epitope-specific repertoire, rendering the host at an increased risk of infection.22,23,25-27 In keeping with this postulate, we provide evidence of restricted (9%-20%) and monoclonal (15%-50%) T-cell responses in the various subgroups studied. T-cell clonal selection might take place locally as a result of a reaction to LPD; however, given the well-reported evidence of restricted T-cell responses associated with aging and other immunosuppression, it is likely that these restricted populations antedate the development of the LPD and stem from an underlying deficiency in T-cell function.
Cases of RH and EBVMCU and a small number of Poly-N cases exhibited a high degree of spontaneous regression. In particular, the EBVMCU cases were characterized by a waxing and waning course. Such a clinical behavior suggests a possibility of partial and spontaneously reversible reduction in immunosurveillance over EBV. No specific pathologic clues could be identified to explain this phenomenon. The features of EBVMCU showed some resemblance to what has been termed EBV-associated genital ulcer, which, interestingly, is usually associated with acute EBV infection in young patients. These lesions characteristically heal spontaneously over a period of 2 to 3 weeks as the EBV-associated immune response becomes effective.28
Clonal IG gene rearrangements are variably present in immunosuppression-associated LPDs, and emergence of a B-cell clone may be an indication of progression.29 The present series showed PCR evidence of B-cell clonality in 16%-63% of cases, with statistically significant differences among the groups (P < .005). Clonality was rarely found in RH and was most frequent in Poly-N and DLBCL. In this context, it is notable that age-related changes similar to those that affect T cells have been detected in B cells as well, with a dramatic collapse in B-cell repertoire diversity and clonal B-cell expansion.30 This may explain the evidence of clonal B cells in several cases of RH.
To date, genetic data on EBV+ DLBCL are limited to a single case with t(9;14)(p13;q32) involving PAX5.17 This translocation, however, has been found in a range of B-cell lymphomas and thus is not specific for EBV+ DLBCL.15 Interphase analysis of 16 cases detected rearrangement of IGH@ in 3 cases (19%), which confirmed a recurrent presence of IGH@-mediated translocations in EBV+ DLBCL. Involvement of PAX5 in these cases was excluded by FISH. Further studies are needed to identify the targeted partner genes, which likely play an important role in the pathogenesis of these lymphomas.
Treatment for AR-EBVLPD published to date has been conventional chemotherapy, radiotherapy, or both used for EBV− DLBCL, with a poor overall response rate of 66% and median survival of 2 years.6,11 Patients in the present study received comparable treatment, although because of the consultation nature of the series, conclusions are limited regarding treatment efficacy. Patients with Poly-N and DLBCL had a similarly low CR rate of 60% and 55%, with an overall survival of 24 and 25 months, respectively. Because the presumed underlying pathogenetic mechanism of AR-EBVLPD is immunosenescence, management approaches in the future might include restoration of immunologic control over EBV. Cultured autologous and pools of allogeneic EBV-specific cytotoxic T cells have been successful in controlling EBV-driven LPDs in a variety of immunosuppression settings.4,31,32 Similarly, IFN-α2b has been used in the management of lymphomatoid granulomatosis and may enhance immune function.33 Our follow-up data identified use of chemotherapy or radiation in 21% of cases of RH and 36% of EBVMCU cases. Given the high rate of spontaneous resolution and indolent, relapsing, and remitting disease in this subset, recognition and careful selection of these cases might facilitate more conservative management.
The pathologic spectrum of DLBCL observed in AR-EBVLPD is broad. A subset of cases (30%) displayed “conventional” morphology. Without EBER testing, these cases might not be recognized as AR-EBVLPD. More typical were cHL-like features, prominent angioinvasion, and marked pleomorphism. Other morphologic variants that are part of the AR-EBVLPD spectrum include cases resembling T-cell/histiocyte-rich large B-cell lymphoma, previously found to be EBV+ in a small subset of cases,34,35 and plasmablastic lymphoma, also found to be associated with a range of immunosuppression settings, including old age.16 The spectrum of lesions observed in AR-EBVLPD in many respects is analogous to that seen in the posttransplantation setting, with progression in some cases from Poly-N or Poly-E to DLBCL. Similar progression has been noted in isolated reports,36 with the suggestion that plasma cell hyperplasia might be the earliest disease manifestation.37
The classification of AR-EBVLPDs is not always straightforward. The border between reactive lesions and lymphoma can be imprecise, and separation between the different AR-EBVLPD subtypes can be problematic. Most cases diagnosed as RH lacked clonal IG rearrangements, which indicates that the identification of a clonal B-cell population favors a more aggressive process. In addition, EBV+ LPDs in general often harbor immunoblasts that resemble HRS cells. Such cells were particularly prominent in the polymorphic and DLBCL cases. The HRS-like cells showed a reduced level of expression of CD20 and, in the cases tested, had retained positivity for PAX5 and Oct-2. Although CD30 was uniformly positive, coexpression of CD15 was variable and seen in up to 62% of cases. These features raise the differential of cHL.7,38 In the series from Oyama et al6,11 and Asano et al,7 CD15 was used as a major criterion, in that no case of AR-EBVLPD was CD15+. The present data suggest that CD15 expression can clearly occur in EBV+ lesions outside the realm of cHL and that one must be cautious in considering the presence or absence of CD15 as a sole discriminatory factor.10 Furthermore, cHL seldom involves extranodal sites; in particular, primary mucosal or cutaneous involvement is considered exceptionally rare, if it occurs at all.39 Interestingly, of the 108 cases of cHL reported by Asano et al7 in patients > 50 years of age, 8 presented in unusual extranodal sites, including skin (2), gastrointestinal tract (4), and lung (2). In the setting of a lymph node biopsy, a useful feature that favored AR-EBVLPD was the wide range in cell size and morphology of the EBV+ cells as highlighted by CD20, CD30, and EBER. EBER also highlighted a greater number and range of positive cells than seen in EBV+ cHL. Another useful feature against the diagnosis of cHL is a range of cells with plasmacytoid morphology similar to that seen in the polymorphic variant of PTLD. Cases with evidence of T-cell clonality pose a differential diagnosis with angioimmunoblastic T-cell lymphoma, a tumor that typically contains numerous EBV+ B cells. Diagnosis of angioimmunoblastic T-cell lymphoma relies on identification of T-cell atypia in the appropriate immunophenotypic context (CD10/CXCL13/PD1+ T cells as well as expanded follicular dendritic cells).
In summary, AR-EBVLPD in the Western population represents a wide pathologic spectrum that includes reactive hyperplasia, nodal and extranodal polymorphic LPDs, and DLBCL, with varied morphologic appearances. The range of clinical behavior is wide, with polymorphic PTLD-like lesions and DLBCL cases having poor survival. The histologic types are highly predictive of outcome, and small-volume disease, particularly in mucosal sites and skin (EBVMCU), is compatible with good prognosis. Analysis of T-cell clonality by PCR highlights restricted or oligoclonal patterns, which suggests a reduced T-cell repertoire as the underlying pathogenetic mechanism, secondary to immunosenescence. The results of the present study advocate for routine EBER testing in patients older than 50 years of age who are suspected to have an aggressive B-cell lymphoma and those presenting with atypical nodal, mucosal, or cutaneous B-cell LPDs.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
We acknowledge Ursula Pluys for technical support of the FISH analysis.
This work was supported by the intramural program of the Center for Cancer Research, National Cancer Institute. S.D.D. received financial support from the British Division of the International Academy of Pathology and British Society for Hematology.
National Institutes of Health
Authorship
Contribution: S.D.D contributed to the study design, performed the research, analyzed data, and wrote the paper; G.V. and M.R. conducted the PCR analyses; S.P. contributed to the study design and provided data; I.W. conducted the FISH analysis; J.A.S. identified cases and collated data; R.K.H. performed the statistical analysis; and E.S.J. designed the project, reviewed, validated, and analyzed data, and wrote and edited the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for J.A.S. is Genzyme Genetics, Hematopathology, New York, NY. The current affiliation for G.V. is Department of Pathology, Loyola University Medical Center, Maywood, IL.
Correspondence: Dr Elaine S. Jaffe, Bldg 10, Rm 2B 42, MSC-1500, 10 Center Dr, National Institutes of Health, Bethesda, MD 20892; e-mail: elainejaffe@nih.gov.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal