

Abstract
Chronic lymphocytic leukemia (CLL) cell migration into lymphoid tissues is an important aspect of the pathobiology of this disease. Here, we investigated the role of ephrin-A4 (EFNA4) in the transendothelial migration (TEM) capacity of CLL and normal B cells through interacting with endothelial EphA2 (erythropoietin-producing hepatocellular carcinoma). CLL cells showed a remarkable impairment in the adhesion to and transmigration through human umbilical vein endothelial cell (HUVEC) monolayers, correlating with their higher EFNA4 expression. In vitro, TEM was mediated by EFNA4 binding to endothelial EphA2 receptor, which is highly expressed in tumor necrosis factor-α–activated HUVECs as well as in the CD31+ endothelial cells of human lymph nodes. The pretreatment of CLL cells with EphA2 homodimers further impaired their adhesion to and transmigration through HUVEC monolayers, whereas pretreatment of HUVECs with EFNA4 homodimers improved those phenomena in both CLL and normal B cells, suggesting that EFNA4 signaling negatively contributed to TEM. In fact, EFNA4 signaling into CLL cells significantly reduced their adhesion to intercellular adhesion molecule 1, vascular cell adhesion molecule 1, and several extracellular matrix molecules and impaired CCL-19–mediated TEM and chemotaxis. Our results suggest that EFNA4-EphA2 interactions are involved in CLL cell trafficking between blood and the tissues and therefore may become a therapeutic target in the future.
Introduction
Chronic lymphocytic leukemia (CLL) is characterized by the progressive accumulation of CD5+ leukemia B cells in the peripheral blood (PB), bone marrow, and lymphoid tissues.1,2 CLL cells continuously traffic between these tissue compartments and form proliferation centers or “pseudofollicles” within the infiltrated lymphoid tissues, likely contributing to tumor growth and survival,3,4 but these issues are still controversial and not fully clear. Thus, a central aspect of the pathophysiology of CLL is the knowledge of the mechanism/s governing CLL-cell extravasation into lymphoid tissues because interfering with this process might be beneficial for disease management. The study in CLL cells of the common molecular mechanisms of lymphocyte entry into lymph nodes (LNs), including chemokine receptors like CCR7, CXCR4, or CXCR5 or cell-adhesion molecules (CAMs) of the integrin family,5-9 has revealed, but not conclusively demonstrated, partial associations between the expression of some of these molecules and a differential migration capacity that could be associated to the occurrence of clinical lymphadenopathy.5,6,8 Finally, several lines of evidence have suggested that CLL cells could have an altered capacity to migrate into the different tissue compartments.10-13 Hence, further work is necessary to unravel the mechanisms of CLL-cell extravasation into tissues through the definition of new molecular players.14
CLL cells differentially express some members of the Eph receptor tyrosine kinase family, partially correlating with clinical features.15 An inverse association between the expression levels of ephrin-A4 (EFNA4) and the presence of clinical lymphadenopathy was found,15 suggesting that EFNA4 could play a functional role in the extravasation capacity of CLL cells.
Eph receptor tyrosine kinases and their membrane-bound ligands, the ephrins (EFNs), are largely known for their role in regulating cell shape and attachment through the modulation of cell adhesion and migration.16-22 They are classified into 2 families, EphA (9 members in humans) and EphB (6 members), depending on the similarity within each group of the extracellular domain sequences and on their affinity for binding EFNs either of type A (5 members), which are glycosylphosphatidylinositol-anchored proteins to the cell membrane, or of type B (3 members), which have a single transmembrane domain.16-19
An essential property of Eph-EFN interaction is that it can result in bidirectional signaling into both the Eph- (forward signaling) and the EFN-expressing cells (reverse signaling).16-19,23,24 The degree of Eph/EFN aggregation during interaction may affect signal strength, leading to opposed cell outcomes, such as adhesion or repulsion between the interacting cells,16-19,23 through modulating and being modulated by other adhesion molecules, including integrins.17
On the basis of these data, we investigated the role played by EFNA4 in the CLL-cell traffic through tissues. We examined whether EFNA4 mediates the adhesion and/or transendothelial migration (TEM) of CLL cells to endothelial cells, thus modulating their capacity for extravasation.
Methods
Human samples
All patients with CLL provided written informed consent to their involvement in the study, which was approved by the Ethics and Research Committees of our participating institutions in accordance with the principles of the Declaration of Helsinki. Patients were untreated and diagnosed according to standard morphologic and immunophenotypic criteria (supplemental Table 1, available on the Blood website; see the Supplemental Materials link at the top of the online article).
Heparanized blood samples were centrifuged onto Histopaque 1.077 cushion (Sigma-Aldrich) to obtain PB mononuclear cells and were then depleted of most T cells by rosetting with 2-amino-ethyl-thio-isouronium bromide hydrobromide (Sigma-Aldrich)–treated sheep erythrocytes (Durviz). Monocytes (CD14+), residual T and natural killer cells (CD2+), and granulocytes (CD13+) were depleted by preincubating the cell suspensions with monoclonal antibodies (mAbs) for the indicated antigens (BD Biosciences), followed by secondary species-specific Abs conjugated to MACS (Miltenyi Biotec) and separated through an AutoMACS cell separator (Miltenyi Biotec). The purities of CLL or normal B-cell enrichments were 98% or greater.
To isolate CD31-expressing endothelial cells from LNs, cell suspensions were prepared through mechanical disruption followed by collagenase digestion (Collagenase Type IV, Invitrogen; 400 U/mL, at 37°C, 1 hour) of fresh LN biopsies (4 CLL lymphadenopathies, 2 reactive LN and 2 normal sentinel LN from patients with breast cancer). T cells were depleted as discussed previously and CD5-, CD19-, CD13-, or CD14-expressing cells through magnetic beads (Dynabeads; Invitrogen). Finally, a positive selection step of CD31-expressing cells was performed with an anti-human CD31 mAb (BD Biosciences) and MACS-coupled secondary Abs (Miltenyi Biotec).
Reverse transcription–polymerase chain reaction analysis of Eph receptor mRNA expression
Total RNA was isolated from cell lysates (Tri-Reagent; Sigma-Aldrich) following the manufacturer's instructions. A total of 2 μg of total RNA was reverse-transcribed into cDNA (Superscript III; Invitrogen). Polymerase chain reaction (PCR) amplifications were performed with an AmpliTaq Gold polymerase (Invitrogen) in a PCR thermal cycler (Eppendorf; see the supplemental Methods section for primers).
After PCR amplification (5 minutes at 93°C initial step, followed by 40 cycles of 15 seconds at 94°C, 30 seconds at 58°C, and 30 seconds at 72°C, followed by 10 minutes at 72°C for final extension), PCR products were analyzed on a 1.2% agarose gel (Biotools) containing ethidium bromide (Sigma-Aldrich) and imaged with a GelDoc UV illuminator (Bio-Rad).
Flow cytometric analyses
Cell suspensions were incubated in cold phosphate-buffered saline (PBS; 0.1% bovine serum albumin [BSA], 2 × 105 cells/50 μL) with the following mAbs: anti-CD19 (fluorescein isothiocyanate [FITC], allophycocyanin, or phycoerythrin [PE]), -CD5 (PECy5), -CD3 (PECy5 or APC); FITC-labeled anti-CD10, -CD11a, -CD44, -CD54; PE-labeled anti-CD23, -CD29, -CCR7; PE-Cy5–labeled anti-CD18, -CD49d; and Alexa Fluor 647–labeled anti-CD31 (all from BD Biosciences). EFNA4 expression was determined with a biotinylated goat anti–human EFNA4 polyclonal Ab (Vitro; R&D Systems) in the presence of purified goat IgG immunoglobulins (Jackson ImmunoResearch Laboratories) followed by streptavidin–Alexa Fluor 488 (Invitrogen). Intracellular anti-EphA2 stainings (goat anti–human EphA2; Santa Cruz Biotechnology) were carried out in permeabilized (Cytofix/Cytoperm buffer; BD Biosciences) human umbilical vein endothelial cell (HUVEC) suspensions, followed by PE-coupled anti–goat secondary Abs (Jackson ImmunoResearch Laboratories). Eph/EFN-Fc binding assays were done by preincubating cell suspensions with purified human IgG Fc fragments (hFc; Jackson ImmunoResearch Laboratories) before the addition of Eph/EFN homodimeric proteins (0.5 μg/106 cells; R&D Systems), followed by a FITC (AbD Serotec) or PE-labeled anti-His mouse mAb (R&D Systems). Data were acquired in a 4-color flow cytometer (FACSCalibur; BD Biosciences) and analyzed with CellQuest software (BD Biosciences).
TEM assays
HUVECs (PromoCell; LabClinics) were grown to confluent monolayer in endothelial-cell culture medium (PromoCell) onto 96-well culture plates (BD Biosciences), overnight activated with 10 ng/mL tumor necrosis factor-α (TNF-α) (PeproTech) in serum-starved medium, and washed in fresh medium for 2 hours. B cells were stained with 2μM carboxyfluorescein succinimidyl ester (CFSE; Invitrogen), added to wells (3 × 105/well), and cocultured for 2 hours. Then, wells were gently and thoroughly washed with warmed medium, thus recovering both nonadhered and weakly adhered B cells (Fraction-1, F-1). A second round of washings with warmed medium containing 5mM EDTA (ethylenediaminetetraacetic acid; Panreac) allowed tightly adhered B cells to be recovered (F-2). Finally, the HUVEC monolayers were detached by treating them with medium containing trypsin/5mM EDTA, thus releasing B cells from beneath the monolayer (F-3). Examination of the plate under the microscope was routinely performed before and after the final washing step to control the integrity and complete detachment, respectively, of the HUVEC monolayer. Absolute B-cell counts were determined by flow cytometry, through gating of CFSE-positive lymphocytes, and counting beads (CountBright absolute counting beads; Invitrogen) following the manufacturer's recommendations, according to the formula:
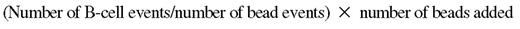
HUVEC-lymphocyte conjugates
TNF-α–activated HUVEC monolayers were stained with PKH-26 fluorescent dye (Sigma-Aldrich). Then, HUVECs were detached by washing with culture medium containing 5mM EDTA. Each sample tube consisted of PKH-26–stained HUVECs (5 × 104) mixed with CFSE (2μM)-stained B cells (2 × 105), spun down and cocultured at 37°C. At the indicated times, cell suspensions were fixed (Cell-fix solution; BD Biosciences) and analyzed by FACS (supplemental Figure 1).
Cell-adhesion assays
Extracellular matrix molecules (ECM) including fibronectin (FN, 10 μg/mL), vitronectin (VN, 10 μg/mL), laminin (10 μg/mL), type I collagen (10 μg/mL; all from BD Biosciences) and hyaluronic acid (10 μg/mL), or recombinant CAMs intracellular adhesion molecule–1 (ICAM-1) and vascular cell adhesion molecule–1 (VCAM-1; 10 μg/mL each, all from R&D Systems) were bound to the flat surface of 96-well culture plates (BD Biosciences) for 2 hours at 37°C. CLL and normal B cells (5 × 105/well, 200 μL final volume) were allowed to adhere for 2 hours. Nonadhered cells were then recovered through agitation and aspiration and counted by FACS (see “TEM assays”).
Chemotaxis assays
Chemotaxis assays were performed in Transwell insert plates (96-well, 5-μm pore size filters; Costar, Corning), with or without a HUVEC monolayer grown onto them. CFSE-stained B cells were added to the upper chamber (5 × 105/well) in the presence of human recombinant chemokines (PeproTech) in the lower chamber (CCL19, 500 ng/mL; CXCL12, 100 ng/mL; CXCL13, 1000 ng/mL). Plates were centrifuged (for 5 seconds at 200g) to spin down the cells onto the filter and migration proceeded for 2 hours in the incubator (37°C, 5% CO2). Migrated cells were recovered from the bottom chamber through washing with 5mM EDTA containing medium to detach cells and counted by FACS (see the section “TEM assays”).
Confocal microscopy studies
Fluorescence images were acquired in a confocal microscope (TCS-SP2 AOBS spectral detection system coupled to an IRE-2 Leica inverted microscope, Leica). Sequential acquisition scanning was carried out by line-averaging (×8) and frame accumulation (×2) at 1024 × 1024 pixel resolution, in a 22°C coiled darkroom. Where indicated, Z-series confocal images (0.1-μm Z-steps) were obtained from apical to basal sides of HUVECs. Colocalization plots and pseudocolor projections of z-stacks were created with LCS software (Leica). Quantification of images was performed with MetaMorph (Molecular Devices; see supplemental Methods).
All incubations with Abs (0.1 μg/50 μL in PBS/BSA) were done in a humidified chamber. Primary Abs were rabbit anti-EphA2, -EphA3, -EphA4 (Santa Cruz Biotechnology) and goat anti-EphA8 (R&D Systems) polyclonal Abs; and mouse anti-CD19, Alexa Fluor 647 anti-CD31, Alexa Fluor 488 anti–ICAM-1, and Alexa Fluor 647 anti–VCAM-1 mAbs (BD Biosciences). Preadsorbed species-specific secondary Abs (Invitrogen) were Alexa Fluor 546–coupled donkey anti–goat or anti–rabbit IgGs and Alexa Fluor 488–coupled donkey anti–mouse IgGs.
Immunofluorescence stainings of tissue sections
Eight-micrometer LN tissue sections (Leica cryocutter at −24°C) were air-dried overnight, fixed for 10 minutes in acetone (Panreac), and air-dried for 1 hour. Prehydrated tissue sections (PBS/0.5% BSA) were then incubated with anti-EphA2, -EphA3, -EphA4, or -EphA8 and anti-CD19 primary Abs followed by Alexa Fluor 546– and Alexa Fluor 488–labeled secondary Abs. After washing, sections were incubated with purified mouse IgGs followed by Alexa Fluor 647–coupled anti-CD31 mAb.
Immunofluorescence staining of TEM assays and colocalization studies
TNF-α–activated HUVEC monolayers were grown onto fibronectin-coated (10 μg/mL) 12-well glass chamber slides (Lab-Tek chamber slides; Nunc). For TEM assays, nonadhered B cells were removed by aspiration, slides fixed (CellFix solution; BD Biosciences), and incubated with anti-EphA2 and Alexa Fluor 488 anti—ICAM-1 primary Abs, then with Alexa Fluor 546–labeled secondary Ab.
For colocalization studies, HUVEC monolayers were cultured in the incubator (37°C, 5% CO2) with fluorescent preclustered forms of either EFNA4-Fc (0.5 μg/106 cells) or of anti–ICAM-1 mAb (0.1 μg/106 cells). Cultures were fixed (CellFix; BD Biosciences) at 15, 30, or 60 minutes. As appropriate, fluorescent EFNA4-Fc treatments were incubated with either anti-EphA2 or -EphA4 primary Abs followed by Alexa Fluor 546 secondary Ab or with Alexa Fluor 488 anti–ICAM-1, Alexa Fluor 647 anti–VCAM-1, and anti-EphA2 primary Abs followed by Alexa Fluor 546 secondary Ab. Anti–ICAM-1 treatments were stained with Alexa Fluor 647 anti–VCAM-1 and anti-EphA2 primary Ab followed by Alexa Fluor 546 secondary Ab.
Statistics
Data are mean values plus or minus SD or SEM from triplicate experiments. SPSS 15.0 for Windows was used for statistical analyses. Mean values were compared by 1- or 2-sample 2-tailed Student t test. Significance was recognized at P values less than .001, .005, or .05.
Results
The CD31+ vascular endothelia of CLL and normal LNs express the EphA2 receptor for EFNA4
As determined by flow cytometry, PB CLL cells show an increased expression of EFNA4 compared with normal B cells (MFI 30.56 ± 15.31 and 8.04 ± 0.23, respectively; Table 1), inversely correlating with clinical lymphadenopathy (MFI 21.11 ± 7.99 and 41.60 ± 14.66, P < .01, with and without lymphadenopathy, respectively; Table 1), in keeping with our previous results.15
EFNA4, cell adhesion molecules, and CCR7 expressions of CLL cells in relation to clinical stage and lymphadenopathy
| Case no. | Stage (Rai)* | Lymphadenopathy | EFNA4 | CD62L | CD44 | CD11a (LFA1α) | CD18 (β2) | CD49d (VLA-4 α) | CD29 (β1) | CCR7 |
|---|---|---|---|---|---|---|---|---|---|---|
| CLL-1 | Lo | No | 37.23 | 89.44 | 166.23 | 7.64 | 10.63 | 13.91 | 8.26 | 4.07 |
| CLL-2 | Lo | No | 32.58 | 18.96 | 144.93 | 14.98 | 15.46 | 45.06 | 14.01 | 6.73 |
| CLL-3 | Lo | No | 52.36 | 48.81 | 113.39 | 36.41 | 35.25 | 20.62 | 15.81 | 6.35 |
| CLL-4 | Lo | No | 65.56 | 121.52 | 929.44 | 14.62 | 22.73 | 29.89 | 11.60 | 39.23 |
| CLL-5 | Int | Yes | 14.95 | 859.23 | 1376.01 | 24.48 | 79.77 | 133.06 | 15.40 | 24.31 |
| CLL-6 | Int | Yes | 15.23 | 181.91 | 924.52 | 11.40 | 40.72 | 123.45 | 17.84 | 12.56 |
| CLL-7 | Int | Yes | 19.98 | 583.33 | 741.23 | 32.12 | 74.90 | 20.33 | 12.39 | 15.23 |
| CLL-8 | Int | Yes | 23.75 | 706.02 | 939.25 | 13.86 | 16.51 | 29.63 | 18.24 | 14.75 |
| CLL-9 | Hi | Yes | 25.56 | 562.23 | 725.23 | 10.67 | 9.83 | 20.25 | 13.27 | 11.70 |
| CLL-10 | Hi | Yes | 12.65 | 868.62 | 441.25 | 10.87 | 9.08 | 22.52 | 14.01 | 10.76 |
| CLL-11 | Hi | Yes | 35.62 | 444.02 | 333.25 | 13.58 | 10.32 | 12.48 | 18.84 | 8.97 |
| CLL-12 | Hi | No | 36.26 | 649.23 | 689.52 | 19.05 | 11.11 | 7.08 | 25.02 | 10.11 |
| CLL-13 | Hi | No | 25.63 | 1740.21 | 1140.32 | 14.97 | 9.10 | 7.20 | 55.85 | 12.25 |
| Case no. | Stage (Rai)* | Lymphadenopathy | EFNA4 | CD62L | CD44 | CD11a (LFA1α) | CD18 (β2) | CD49d (VLA-4 α) | CD29 (β1) | CCR7 |
|---|---|---|---|---|---|---|---|---|---|---|
| CLL-1 | Lo | No | 37.23 | 89.44 | 166.23 | 7.64 | 10.63 | 13.91 | 8.26 | 4.07 |
| CLL-2 | Lo | No | 32.58 | 18.96 | 144.93 | 14.98 | 15.46 | 45.06 | 14.01 | 6.73 |
| CLL-3 | Lo | No | 52.36 | 48.81 | 113.39 | 36.41 | 35.25 | 20.62 | 15.81 | 6.35 |
| CLL-4 | Lo | No | 65.56 | 121.52 | 929.44 | 14.62 | 22.73 | 29.89 | 11.60 | 39.23 |
| CLL-5 | Int | Yes | 14.95 | 859.23 | 1376.01 | 24.48 | 79.77 | 133.06 | 15.40 | 24.31 |
| CLL-6 | Int | Yes | 15.23 | 181.91 | 924.52 | 11.40 | 40.72 | 123.45 | 17.84 | 12.56 |
| CLL-7 | Int | Yes | 19.98 | 583.33 | 741.23 | 32.12 | 74.90 | 20.33 | 12.39 | 15.23 |
| CLL-8 | Int | Yes | 23.75 | 706.02 | 939.25 | 13.86 | 16.51 | 29.63 | 18.24 | 14.75 |
| CLL-9 | Hi | Yes | 25.56 | 562.23 | 725.23 | 10.67 | 9.83 | 20.25 | 13.27 | 11.70 |
| CLL-10 | Hi | Yes | 12.65 | 868.62 | 441.25 | 10.87 | 9.08 | 22.52 | 14.01 | 10.76 |
| CLL-11 | Hi | Yes | 35.62 | 444.02 | 333.25 | 13.58 | 10.32 | 12.48 | 18.84 | 8.97 |
| CLL-12 | Hi | No | 36.26 | 649.23 | 689.52 | 19.05 | 11.11 | 7.08 | 25.02 | 10.11 |
| CLL-13 | Hi | No | 25.63 | 1740.21 | 1140.32 | 14.97 | 9.10 | 7.20 | 55.85 | 12.25 |
Numbers are mean fluorescence intensity (MFI) values as determined by flow cytometry. EFNA4 (MFI) of normal B cells: 8.04 ± 0.23 (mean ± SEM from 3 different samples).
CLL indicates chronic lymphocytic leukemia.
Clinical stage of untreated CLL patients (modified Rai staging) at the moment of blood sampling: low (Lo, Rai 0), intermediate (Int, Rai I-II) and high risk (Hi, Rai III-IV).
We analyzed the expression of EphA receptors in vascular endothelial cells from the enlarged LNs of 3 patients with CLL. As determined by flow cytometry, most (98% ± 4.66%) CD31+CD5−CD19− cells from CLL LNs strongly bound EFNA4-Fc proteins compared with the other LN-cell subpopulations studied (Figure 1A). RT-PCR analyses of EphAs mRNA expression in isolated CD31+CD2−CD5−CD19−CD13−CD14− LN cells (3 CLL lymphadenopathies, 2 reactive LN, and 2 otherwise-normal sentinel LN), showed a high expression of EphA2 and, to a lesser extent, of EphA3, EphA4, EphA5, and EphA8 mRNAs (Figure 1B). Immunofluorescence studies in LN tissue sections confirmed a preferential expression of EphA2 by the CD31+ vascular endothelium (Figure 1C), including high endothelial venules (Figure 1D). By contrast, EphA3, EphA4, and EphA8 were rarely expressed by the CD31+ vascular structures (Figure 1C). These results supported the thesis that EphA2-EFNA4 interactions could take place during TEM of CLL and normal B cells.

EphA2 is the main EFNA4 receptor found in the CD31+ vascular endothelium of human LN. (A) Flow cytometric analyses of EFNA4-Fc binding by LN cells from patients with CLL (a representative experiment is shown, CLL LN no. 2). Cell suspensions (3 × 105/sample tube) from fresh CLL LN biopsies were preincubated with hFc before the addition of poly-His–tagged EFNA4-Fc homodimers (0.5 μg/106 cells), then followed by an anti His-FITC mAb. The percentage of EFNA4-Fc binding cells (black profiles) was analyzed within gated subpopulations according to CD5, CD19, and CD31 costainings (empty profiles: control anti-His–FITC stainings). (B) RT-PCR analyses of EphA mRNAs expression of enriched CD2− CD5− CD19− CD13− CD14− CD31+ LN cells from CLL patients or control subjects (≥ 95% purity, 2-5 × 105 cells per LN fragment). (C-D) Immunofluorescence analysis of EphA receptor expression on LN tissue sections. LN cryosections (8 μm thick) were immunostained for detection of EphA2, EphA3, EphA4, or EphA8 (Alexa Fluor 546, green), CD31 (Alexa Fluor 647, red), and CD19 (Alexa Fluor 488, blue). Objectives: 20× multi-immersion, 1.20 NA. (D) High magnification images (objective: 63× oil immersion, 1.40 NA) of CLL or normal LN showing expression of EphA2 (green) in the high endothelial venules (CD31+ structures, red; CLL LN no. 2 in panels C-D; Normal reactive LN in panel D). Fluorescence images were acquired with a confocal microscope (Leica TCS-SP2 AOBS).
EphA2 is the main EFNA4 receptor found in the CD31+ vascular endothelium of human LN. (A) Flow cytometric analyses of EFNA4-Fc binding by LN cells from patients with CLL (a representative experiment is shown, CLL LN no. 2). Cell suspensions (3 × 105/sample tube) from fresh CLL LN biopsies were preincubated with hFc before the addition of poly-His–tagged EFNA4-Fc homodimers (0.5 μg/106 cells), then followed by an anti His-FITC mAb. The percentage of EFNA4-Fc binding cells (black profiles) was analyzed within gated subpopulations according to CD5, CD19, and CD31 costainings (empty profiles: control anti-His–FITC stainings). (B) RT-PCR analyses of EphA mRNAs expression of enriched CD2− CD5− CD19− CD13− CD14− CD31+ LN cells from CLL patients or control subjects (≥ 95% purity, 2-5 × 105 cells per LN fragment). (C-D) Immunofluorescence analysis of EphA receptor expression on LN tissue sections. LN cryosections (8 μm thick) were immunostained for detection of EphA2, EphA3, EphA4, or EphA8 (Alexa Fluor 546, green), CD31 (Alexa Fluor 647, red), and CD19 (Alexa Fluor 488, blue). Objectives: 20× multi-immersion, 1.20 NA. (D) High magnification images (objective: 63× oil immersion, 1.40 NA) of CLL or normal LN showing expression of EphA2 (green) in the high endothelial venules (CD31+ structures, red; CLL LN no. 2 in panels C-D; Normal reactive LN in panel D). Fluorescence images were acquired with a confocal microscope (Leica TCS-SP2 AOBS).
An altered TEM of CLL cells through HUVEC monolayers is mediated by EphA2-EFNA4 interactions
To ascertain a plausible role of EphA2-EFNA4 interaction in the TEM of CLL cells or normal B cells, we carried out in vitro transmigration assays with TNF-α–activated HUVEC monolayers, which strongly expressed EphA2 and bound EFNA4-Fc fusion protein (Figure 2A). CLL cells from 7 of the 13 different CLL patients studied (nos. 1, 3, 4, 5, 8, 10, and 13), differing in EFNA4 expression (Table 1), showed a poorer adhesion and TEM than normal B cells (Table 2). As determined by confocal microscopy analysis of similarly performed TEM experiments, endothelial EphA2 was largely concentrated in the cell contacts with B cells (Figure 2B), suggesting that EphA2-EFNA4 interactions can take place in the adhesion between both cell types.

EphA2-EFNA4 interactions can take place in the adhesion between HUVECs and B cells during TEM. (A) Flow cytometry analysis of EFNA4-Fc binding (top histogram, gray) or EphA2 expression (bottom histogram, gray) of TNF-α–activated HUVECs (>95% CD31, CD54 double positive, dot-plot; black histogram: control secondary PE-Abs; white: unstained cells). (B) CLL cells or normal B cells were cocultured for 2 hours with TNF-α–activated HUVEC monolayers, the nonadhered cells were washed out, and slides were fixed and immunostained for EphA2 (Alexa Fluor 546, red, bottom) and ICAM-1 (Alexa Fluor 488, green, top; blue, Hoechst 33 342 counterstained cell nuclei; confocal microscope: Leica TCS-SP2 AOBS; objectives: 20× oil-immersion, 1.20 NA). Note the high number of normal B cells transmigrated (dark appearance) compared with cultures containing CLL cells in the phase-contrast images. (C) Flow cytometric analyses of EphA2-Fc binding by CLL cells directly incubated with EphA2-Fc homodimers (gray, top) or after preincubation with 2 different doses of an anti-EFNA4 polyclonal Ab (0.10 or 0.20 μg/106 cells, middle and bottom, respectively; white, control anti-His FITC; black, control unstained cells). A representative experiment is shown (CLL no. 1). Values are mean fluorescence intensity (MFI; mean ± SEM) from triplicates of the same sample. (D) TNF-α–activated HUVEC monolayers were cultured for 60 minutes with fluorescent preclustered EFNA4-Fc complexes (red). After cells fixation, slides were immunostained with anti-EphA2 or -EphA4 Abs (Alexa Fluor 488, green). Colocalization plots (bottom) show that EFNA4-Fc preferentially binds to EphA2 (ROI indicates region of interest). Leica TCS-SP2 AOBS confocal microscope; objective: 63× oil immersion, 1.40 NA.
EphA2-EFNA4 interactions can take place in the adhesion between HUVECs and B cells during TEM. (A) Flow cytometry analysis of EFNA4-Fc binding (top histogram, gray) or EphA2 expression (bottom histogram, gray) of TNF-α–activated HUVECs (>95% CD31, CD54 double positive, dot-plot; black histogram: control secondary PE-Abs; white: unstained cells). (B) CLL cells or normal B cells were cocultured for 2 hours with TNF-α–activated HUVEC monolayers, the nonadhered cells were washed out, and slides were fixed and immunostained for EphA2 (Alexa Fluor 546, red, bottom) and ICAM-1 (Alexa Fluor 488, green, top; blue, Hoechst 33 342 counterstained cell nuclei; confocal microscope: Leica TCS-SP2 AOBS; objectives: 20× oil-immersion, 1.20 NA). Note the high number of normal B cells transmigrated (dark appearance) compared with cultures containing CLL cells in the phase-contrast images. (C) Flow cytometric analyses of EphA2-Fc binding by CLL cells directly incubated with EphA2-Fc homodimers (gray, top) or after preincubation with 2 different doses of an anti-EFNA4 polyclonal Ab (0.10 or 0.20 μg/106 cells, middle and bottom, respectively; white, control anti-His FITC; black, control unstained cells). A representative experiment is shown (CLL no. 1). Values are mean fluorescence intensity (MFI; mean ± SEM) from triplicates of the same sample. (D) TNF-α–activated HUVEC monolayers were cultured for 60 minutes with fluorescent preclustered EFNA4-Fc complexes (red). After cells fixation, slides were immunostained with anti-EphA2 or -EphA4 Abs (Alexa Fluor 488, green). Colocalization plots (bottom) show that EFNA4-Fc preferentially binds to EphA2 (ROI indicates region of interest). Leica TCS-SP2 AOBS confocal microscope; objective: 63× oil immersion, 1.40 NA.
TEM of CLL cells and normal B cells through HUVEC monolayers and the effect of EphA2-Fc or EFNA4-Fc addition
| Sample/fraction no. | Treatment, % | ||
|---|---|---|---|
| hFc control | EFNA4-Fc | EphA2-Fc | |
| CLL-1 | |||
| F1 | 96.49 ± 0.11 | 76.91 ± 0.10* | 99.21 ± 0.03* |
| F2 | 3.35 ± 0.05 | 22.94 ± 0.09* | 0.78 ± 0.04* |
| F3 | 0.16 ± 0.05 | 0.16 ± 0.01 | 0.02 ± 0.01 |
| CLL-3 | |||
| F1 | 94.66 ± 0.91 | 80.73 ± 1.46† | 96.58 ± 0.31 |
| F2 | 3.72 ± 0.38 | 17.10 ± 1.20‡ | 2.60 ± 0.07 |
| F3 | 1.63 ± 0.53 | 2.17 ± 0.25 | 0.82 ± 0.24 |
| CLL-4 | |||
| F1 | 96.47 ± 0.25 | 73.35 ± 0.15* | 98.93 ± 0.08† |
| F2 | 3.20 ± 0.26 | 26.21 ± 0.15* | 1.06 ± 0.08† |
| F3 | 0.32 ± 0.01 | 0.45 ± 0.00‡ | 0.00 ± 0.00* |
| CLL-5 | |||
| F1 | 94.47 ± 0.08 | 65.80 ± 0.26* | 98.94 ± 0.22‡ |
| F2 | 5.30 ± 0.06 | 33.81 ± 0.26* | 1.06 ± 0.23† |
| F3 | 0.24 ± 0.02 | 0.40 ± 0.01† | 0.00 ± 0.00† |
| CLL-8 | |||
| F1 | 82.62 ± 2.13 | 62.32 ± 1.54† | 95.12 ± 0.95† |
| F2 | 11.29 ± 1.47 | 22.68 ± 0.45† | 3.11 ± 0.64† |
| F3 | 6.10 ± 0.66 | 15.00 ± 1.09† | 1.78 ± 0.30† |
| CLL-10 | |||
| F1 | 75.49 ± 0.42 | 48.35 ± 0.85* | 89.24 ± 0.16* |
| F2 | 16.10 ± 0.21 | 35.83 ± 0.58* | 4.32 ± 0.02* |
| F3 | 8.41 ± 0.21 | 15.82 ± 0.27† | 6.45 ± 0.18† |
| CLL-13 | |||
| F1 | 90.83 ± 0.12 | 63.23 ± 0.16* | 98.98 ± 0.00* |
| F2 | 8.26 ± 0.09 | 35.90 ± 0.14* | 1.01 ± 0.01* |
| F3 | 0.91 ± 0.03 | 0.88 ± 0.02 | 0.02 ± 0.01* |
| NBC-1 | |||
| F1 | 50.94 ± 0.08 | 44.78 ± 1.08† | 51.73 ± 0.37 |
| F2 | 27.25 ± 0.35 | 28.51 ± 0.70 | 22.43 ± 0.61† |
| F3 | 21.80 ± 0.43 | 26.72 ± 0.38† | 25.85 ± 0.23† |
| NBC-2 | |||
| F1 | 60.59 ± 0.56 | 43.17 ± 0.25* | 55.62 ± 0.54† |
| F2 | 26.45 ± 0.61 | 40.90 ± 0.15* | 18.89 ± 0.16‡ |
| F3 | 12.95 ± 0.05 | 15.94 ± 0.10* | 25.50 ± 0.70‡ |
| Sample/fraction no. | Treatment, % | ||
|---|---|---|---|
| hFc control | EFNA4-Fc | EphA2-Fc | |
| CLL-1 | |||
| F1 | 96.49 ± 0.11 | 76.91 ± 0.10* | 99.21 ± 0.03* |
| F2 | 3.35 ± 0.05 | 22.94 ± 0.09* | 0.78 ± 0.04* |
| F3 | 0.16 ± 0.05 | 0.16 ± 0.01 | 0.02 ± 0.01 |
| CLL-3 | |||
| F1 | 94.66 ± 0.91 | 80.73 ± 1.46† | 96.58 ± 0.31 |
| F2 | 3.72 ± 0.38 | 17.10 ± 1.20‡ | 2.60 ± 0.07 |
| F3 | 1.63 ± 0.53 | 2.17 ± 0.25 | 0.82 ± 0.24 |
| CLL-4 | |||
| F1 | 96.47 ± 0.25 | 73.35 ± 0.15* | 98.93 ± 0.08† |
| F2 | 3.20 ± 0.26 | 26.21 ± 0.15* | 1.06 ± 0.08† |
| F3 | 0.32 ± 0.01 | 0.45 ± 0.00‡ | 0.00 ± 0.00* |
| CLL-5 | |||
| F1 | 94.47 ± 0.08 | 65.80 ± 0.26* | 98.94 ± 0.22‡ |
| F2 | 5.30 ± 0.06 | 33.81 ± 0.26* | 1.06 ± 0.23† |
| F3 | 0.24 ± 0.02 | 0.40 ± 0.01† | 0.00 ± 0.00† |
| CLL-8 | |||
| F1 | 82.62 ± 2.13 | 62.32 ± 1.54† | 95.12 ± 0.95† |
| F2 | 11.29 ± 1.47 | 22.68 ± 0.45† | 3.11 ± 0.64† |
| F3 | 6.10 ± 0.66 | 15.00 ± 1.09† | 1.78 ± 0.30† |
| CLL-10 | |||
| F1 | 75.49 ± 0.42 | 48.35 ± 0.85* | 89.24 ± 0.16* |
| F2 | 16.10 ± 0.21 | 35.83 ± 0.58* | 4.32 ± 0.02* |
| F3 | 8.41 ± 0.21 | 15.82 ± 0.27† | 6.45 ± 0.18† |
| CLL-13 | |||
| F1 | 90.83 ± 0.12 | 63.23 ± 0.16* | 98.98 ± 0.00* |
| F2 | 8.26 ± 0.09 | 35.90 ± 0.14* | 1.01 ± 0.01* |
| F3 | 0.91 ± 0.03 | 0.88 ± 0.02 | 0.02 ± 0.01* |
| NBC-1 | |||
| F1 | 50.94 ± 0.08 | 44.78 ± 1.08† | 51.73 ± 0.37 |
| F2 | 27.25 ± 0.35 | 28.51 ± 0.70 | 22.43 ± 0.61† |
| F3 | 21.80 ± 0.43 | 26.72 ± 0.38† | 25.85 ± 0.23† |
| NBC-2 | |||
| F1 | 60.59 ± 0.56 | 43.17 ± 0.25* | 55.62 ± 0.54† |
| F2 | 26.45 ± 0.61 | 40.90 ± 0.15* | 18.89 ± 0.16‡ |
| F3 | 12.95 ± 0.05 | 15.94 ± 0.10* | 25.50 ± 0.70‡ |
TEM assays were carried out by coculturing normal B cells or CLL cells with HUVEC monolayers during 2 hours, and nonadhered (F1), adhered (F2), or transmigrating cells (F3) were counted by flow cytometry. The absolute number of cells recovered within each fraction (Fn) was normalized to the total number of them recovered (Fn/F1 + F2 + F3) and expressed as percentages (%). CLL/normal B cells or HUVECs were pretreated with saturating amounts of EphA2-Fc or EFNA4-Fc, respectively. Values are mean ± SD from triplicates. Significant differences between treated and control hFc cultures were calculated by 1-sample, 2-tailed Student t test.
CLL indicates chronic lymphocytic leukemia; F, fraction; hFc, Fc fragment of human IgG immunoglobulin; HUVEC, human umbilical vein endothelial cell; NBC, normal B cell; and TEM, transendothelial migration.
Value of P < .001.
Value of P < .005.
Value of P < .05.
To evaluate the role of this interaction in the TEM assays, HUVECs or B cells were pretreated separately with EFNA4-Fc or EphA2-Fc homodimers, respectively, thus blocking EphA2-EFNA4 interactions whereas cells binding of Eph/EFN-Fc homodimers could induce signaling into them (supplemental Figure 2).25 As determined by flow cytometry, CLL cells from the 7 cases analyzed bound EphA2-Fc homodimers (Figure 2C), which was largely inhibited by preincubating cells with an anti-EFNA4 Ab (Figure 2C). Confocal microscopy studies showed that HUVECs bound EFNA4-Fc preferentially through EphA2 but not EphA4 (Figure 2D), another putative receptor for EFNA4 found in HUVECs (RT-PCR analyses not shown).
EphA2-Fc pretreatment of CLL cells further impaired TEM (Table 2), slightly improving that of normal B cells (Table 2). By contrast, EFNA4-Fc pretreatment of HUVECs reversed the impaired TEM of CLL cells (Table 2), as the number of them adhered increased (Table 2) in correlation with a significant decrease in the nonadhered cells (Table 2), which was accompanied, in 4 of the CLL samples (CLL nos. 4, 5, 8, and 10), by a significant increase in transmigrated cells (Table 2). These results suggested that EFNA4 signaling into the highly positive CLL cells can inhibit TEM, through modulating adhesion and/or transmigration, whereas EphA2 signaling into endothelia promotes this process.
CLL cells have an impaired adhesion to HUVECs that is related to EFNA4 expression
To evaluate whether the adhesion potential of CLL cells to HUVECs can be correlated with clinical-stage and EFNA4 expression, we measured the formation of conjugates between both cell types by flow cytometry (supplemental Figure 1). In control conditions (hFc-only pretreated cells), CLL cells from the 13 patients examined (Table 1) showed a reduced adhesion to HUVECs compared with normal B cells (conjugate percentages at 60 minutes = 8.58 ± 3.76 and 27.27 ± 4.39, respectively; Figure 3A). Among CLL cases, adhesion was significantly greater in advanced disease stages (11.99 ± 1.93 and 9.76 ± 3.40, intermediate or high-risk stages, respectively) than in the low-risk ones (4.55 ± 1.96; Figure 3A) as well as in those cases having lymphadenopathy (11.94 ± 2.17; CLL nos. 5-11) than in those not having it (5.23 ± 1.90; CLL nos. 1-4, 12, and 13; P < .001), inversely correlating with EFNA4 expression (Figure 3B).

CLL cells show an impaired adhesion to HUVECs that can be correlated with EFNA4 expression and EphA2-EFNA4 interactions. (A) The percentage of CLL (nos. 1-13) or normal B cells (3 samples), adhered to HUVECs (conjugates) were analyzed by flow cytometry (supplemental Figure 1) after 60 minutes coculture of PKH26-labeled HUVECs with the CFSE-stained B cells. Data are mean ± SD. (B) Percentage of conjugated CLL cells at 60 minutes relative to EFNA4 expression (MFI, Table 1; r, Pearson correlation coefficient, P < .05). (C) HUVECs or B cells (CLL/normal) were preincubated separately with saturating amounts of EFNA4-Fc or EphA2-Fc homodimers, respectively (0.5 μg/106 cells), then washed and cocultured for 60 minutes, as in panel A. The percentage of conjugates in the treated cocultures was expressed as x-fold change relative to control conditions (hFc; x-coordinate crosses y-coordinate at 1). Most treatments were significantly different from hFc controls (P ≤ .05) except that of normal B cells plus EphA2-Fc (nonsignificant).
CLL cells show an impaired adhesion to HUVECs that can be correlated with EFNA4 expression and EphA2-EFNA4 interactions. (A) The percentage of CLL (nos. 1-13) or normal B cells (3 samples), adhered to HUVECs (conjugates) were analyzed by flow cytometry (supplemental Figure 1) after 60 minutes coculture of PKH26-labeled HUVECs with the CFSE-stained B cells. Data are mean ± SD. (B) Percentage of conjugated CLL cells at 60 minutes relative to EFNA4 expression (MFI, Table 1; r, Pearson correlation coefficient, P < .05). (C) HUVECs or B cells (CLL/normal) were preincubated separately with saturating amounts of EFNA4-Fc or EphA2-Fc homodimers, respectively (0.5 μg/106 cells), then washed and cocultured for 60 minutes, as in panel A. The percentage of conjugates in the treated cocultures was expressed as x-fold change relative to control conditions (hFc; x-coordinate crosses y-coordinate at 1). Most treatments were significantly different from hFc controls (P ≤ .05) except that of normal B cells plus EphA2-Fc (nonsignificant).
These results suggested a role for this molecule in the TEM of CLL cells. In support of this, EphA2-Fc and EFNA4-Fc treatments differentially affected the adhesion of CLL or normal B cells to HUVECs (Figure 3C). Although EphA2-Fc treatment significantly inhibited adhesion of CLL cells (up to 0.2 times relative to hFc conditions; Figure 3C), EFNA4-Fc pretreatment of HUVECs markedly increased it in both B-cell types (up to 2.6 times relative to hFc conditions in CLL no. 3; Figure 3C).
EFNA4 signaling into CLL cells results in a reduced adhesion to different substrates
Upon EphA2-Fc treatment of CLL cells from 7 different patients (CLL nos. 1, 3, 4, 5, 8, 10, and 13), no significant changes of expression were observed in several CAMs examined implicated in the TEM of lymphocytes, including CD62-L (L-selectin), CD11a (αL subunit of integrin LFA-1), CD18 (β2-integrin), CD29 (β1-integrin), or CD49d (α4 integrin, VLA-4), except for down-modulation in the CD44 antigen (Figure 4A).

EFNA4 signaling of CLL cells down-modulates CD44 expression and decreases their adhesion to ECM molecules and ICAM-1 or VCAM-1 CAMs. (A) CLL cells were cultured for 1 hour in the absence (empty profiles) or presence (gray histograms) of saturating amounts (0.5 μg/106 cells) of soluble EphA2-Fc homodimers, then stained with Abs for flow cytometric analyses of CD18, CD29, CD62-L (top), CD11a, CD49d, or CD44 expressions (bottom; black histograms, background staining). A representative experiment is shown. (B) EphA2-Fc–incubated (0.5 μg/106 cells) or hFc-only–treated CLL cells (CLL nos. 1, 3, 4, 5, 8, 10, and 13), corresponding to different EFNA4 expression levels (Table 1), or normal B cells (3 samples), were cultured for 2 hours onto ECM- or CAM-coated culture wells (3 × 105/well). Nonadhered cells were recovered and counted by the use of FACS. A measure of the effect of EphA2-Fc treatment on the cells adhesion was expressed as x-fold change relative to control hFc (number of nonadhered cells recovered in the treated cultures divided by those recovered in the corresponding hFc control ones) and represented against the corresponding EFNA4 expression (MFI) as determined by FACS (r, Pearson correlation coefficient, P < .05). Values are mean from triplicates.
EFNA4 signaling of CLL cells down-modulates CD44 expression and decreases their adhesion to ECM molecules and ICAM-1 or VCAM-1 CAMs. (A) CLL cells were cultured for 1 hour in the absence (empty profiles) or presence (gray histograms) of saturating amounts (0.5 μg/106 cells) of soluble EphA2-Fc homodimers, then stained with Abs for flow cytometric analyses of CD18, CD29, CD62-L (top), CD11a, CD49d, or CD44 expressions (bottom; black histograms, background staining). A representative experiment is shown. (B) EphA2-Fc–incubated (0.5 μg/106 cells) or hFc-only–treated CLL cells (CLL nos. 1, 3, 4, 5, 8, 10, and 13), corresponding to different EFNA4 expression levels (Table 1), or normal B cells (3 samples), were cultured for 2 hours onto ECM- or CAM-coated culture wells (3 × 105/well). Nonadhered cells were recovered and counted by the use of FACS. A measure of the effect of EphA2-Fc treatment on the cells adhesion was expressed as x-fold change relative to control hFc (number of nonadhered cells recovered in the treated cultures divided by those recovered in the corresponding hFc control ones) and represented against the corresponding EFNA4 expression (MFI) as determined by FACS (r, Pearson correlation coefficient, P < .05). Values are mean from triplicates.
Then, we evaluated whether EphA2-Fc–treated CLL cells modulate their adhesion to several of the ligands for the CAMs examined (Figure 4B). EphA2-Fc–treated CLL cells from the 7 patients showed a marked decrease in adhesion to most of the ECM components tested, specially FN, VN, and type I collagen (Figure 4B) but also to ICAM-1 and VCAM-1 (Figure 4B). By contrast, normal B cells did not significantly change their adhesion to these ligands after EphA2-Fc treatment (Figure 4B). These results suggest that EFNA4 signaling plays a cell-autonomous role in the adhesion properties of CLL cells to endothelium, largely inhibiting it, which can be linked to an impaired TEM potential.
EFNA4 signaling into CLL cells inhibits CCL-19 chemotaxis
CLL cells express functional CCR7, CXCR4, and CXCR5 chemokine receptors, which could be implicated in their migration into LN.6-8 Thus, CLL TEM assays were performed in the presence of CCL19, CXCL12, or CXCL13 chemokine gradients and the effect of EphA2-Fc or EFNA4-Fc treatments was evaluated. In control cultures, CLL cells (CLL nos. 1, 3, 4, 5, 8, 10, and 13) showed a better TEM in response to CCL19 chemokine than to CXCL12 or CXCL13 (Figure 5A). EphA2-Fc–treated CLL cells transmigrated significantly less than untreated cells in response to all 3 chemokines (Figure 5A). By contrast, although EFNA4-Fc pretreatment of HUVECs significantly enhanced the CCL19 mediated TEM of CLL cells, it had a negative effect on that induced by CXCL12 or CXCL13 chemokines (Figure 5A).

CCL19-, CXCL12-, or CXCL13-mediated TEM of CLL cells can be modulated by the EphA2-EFNA4 interaction. (A) CLL cells differing in EFNA4 expression (CLL nos. 1, 3, 4, 5, 8, 10, and 13) or normal B cells (3 samples; Table 1), were labeled with CFSE and added to the upper chamber of Transwell inserts (5 × 105/ well), which contained a HUVEC monolayer grown onto them and chemokines added to the lower chamber (CCL19, 500 ng/mL; CXCL12, 100 ng/mL; CXCL13, 1000 ng/mL). HUVECs or CLL/normal B cells were treated separately with EFNA4-Fc or EphA2-Fc homodimers (0.5 μg/106 cells, 30 minutes, 37°C), and chemotaxis was allowed to proceed for 2 hours. The number of cells migrated was determined by FACS. Mean values from triplicate experiments were compared with respect to control conditions (hFc-only–treated cells). The statistically significant differences are indicated by the P value. (B) Chemotaxis assays were performed as in panel A, in the absence of HUVECs and of the corresponding EFNA4-Fc treatment. Experiments were done in triplicate.
CCL19-, CXCL12-, or CXCL13-mediated TEM of CLL cells can be modulated by the EphA2-EFNA4 interaction. (A) CLL cells differing in EFNA4 expression (CLL nos. 1, 3, 4, 5, 8, 10, and 13) or normal B cells (3 samples; Table 1), were labeled with CFSE and added to the upper chamber of Transwell inserts (5 × 105/ well), which contained a HUVEC monolayer grown onto them and chemokines added to the lower chamber (CCL19, 500 ng/mL; CXCL12, 100 ng/mL; CXCL13, 1000 ng/mL). HUVECs or CLL/normal B cells were treated separately with EFNA4-Fc or EphA2-Fc homodimers (0.5 μg/106 cells, 30 minutes, 37°C), and chemotaxis was allowed to proceed for 2 hours. The number of cells migrated was determined by FACS. Mean values from triplicate experiments were compared with respect to control conditions (hFc-only–treated cells). The statistically significant differences are indicated by the P value. (B) Chemotaxis assays were performed as in panel A, in the absence of HUVECs and of the corresponding EFNA4-Fc treatment. Experiments were done in triplicate.
In the absence of HUVEC monolayers, only the CCL19-mediated chemotaxis of CLL cells was significantly inhibited by EphA2-Fc treatment (Figure 5B), suggesting that EFNA4 modulates cell-autonomously the CCR7-mediated chemotaxis.
Normal B cells also showed a significantly increased CCL19-mediated TEM upon EFNA4-Fc treatment of HUVECs (Figure 5A) and, in the absence of endothelial cells, EphA2-Fc treatment slightly inhibited CCL19 chemotaxis (Figure 5B). No significant changes in the TEM or chemotaxis of normal B cells mediated by CXCL12 or CXCL13 chemokines were noted (not shown).
EphA2 clustering on HUVECs recruits ICAM-1 and VCAM-1
EFNA4-Fc–induced relocalization of EphA2 on HUVECs (Figure 2D) could be also associated with an ICAM-1 and VCAM-1 topologic redistribution, both implicated in the lymphocytes' TEM. EFNA4-Fc fluorescent spots formed on the cell surface of cultured HUVEC monolayers, migrated inside the cell, and concentrated perinuclearly in the basal side of HUVECs during a 60-minute culture (Figure 6A). The EphA2 clusterization induced by EFNA4-Fc was accompanied by an ICAM-1 and, to a lesser extent, VCAM-1 sequestration (Figure 6B). In contrast, cross-linking of ICAM-1 on HUVECs did not result in a similar colocalization with EphA2 (Figure 6B), suggesting that ICAM-1 can act independently of EphA2.

EphA2 colocalizes with ICAM-1 and VCAM-1 after in vitro EFNA4-Fc treatment of HUVECs. (A) HUVEC monolayers grown onto glass chamber slides and overnight activated with TNF-α (10 ng/mL) were incubated for 60 minutes with Alexa Fluor 405–EFNA4-Fc protein complexes. After cell fixation of cultures (at 15, 30, or 60 minutes), confocal images were acquired from the apical to the basal side of cells (0.1-μm z-steps; 63× immersion-oil objective). Pseudocolor Z-series projections of image stacks were created (Leica LCS software) to determine the topologic distribution of EFNA4-Fc spots at each time point (left). At each time point, the number of basal and apical EFNA4-Fc spots was determined through image segmentation and analysis (MetaMorph version 7.1; right), according to the color threshold established in the pseudocolor scale bar (red regions, apical; blue regions, basal). Images are from a representative experiment after 60 minutes of culture. (B) HUVECs monolayers grown as in panel A were treated with either EFNA4–Fc protein complexes (preclustered with biotinylated anti-His Ab followed by streptavidin–Alexa Fluor 405) or cross-linking anti–ICAM-1 mAb (anti–ICAM-1 primary Ab preclustered with anti–mouse Alexa Fluor 488 Ab, green). Cultures were fixed after 15, 30, or 60 minutes and immunostained to analyze the colocalization of clustered EphA2 (anti-EphA2 plus Alexa Fluor 546 secondary Ab) with ICAM-1 (Alexa Fluor 488 Ab) or VCAM-1 (Alexa Fluor 633 Ab) or of EphA2 (Alexa Fluor 546) or VCAM-1 (Alexa Fluor 633 Ab) with ICAM-1, respectively. Z-series confocal images were acquired as in panel A. Fluorescent images are from a representative experiment of EFNA4–Fc treatment. Fluorescent blue-red spots, corresponding to EFNA4-Fc aggregated EphA2 (left), were identified as objects through color thresholding (Metamorph software; white regions). The percentage of these regions containing ICAM-1 (green, right), was measured to determine EphA2 colocalization. The graphs represent the results from the corresponding treatments.
EphA2 colocalizes with ICAM-1 and VCAM-1 after in vitro EFNA4-Fc treatment of HUVECs. (A) HUVEC monolayers grown onto glass chamber slides and overnight activated with TNF-α (10 ng/mL) were incubated for 60 minutes with Alexa Fluor 405–EFNA4-Fc protein complexes. After cell fixation of cultures (at 15, 30, or 60 minutes), confocal images were acquired from the apical to the basal side of cells (0.1-μm z-steps; 63× immersion-oil objective). Pseudocolor Z-series projections of image stacks were created (Leica LCS software) to determine the topologic distribution of EFNA4-Fc spots at each time point (left). At each time point, the number of basal and apical EFNA4-Fc spots was determined through image segmentation and analysis (MetaMorph version 7.1; right), according to the color threshold established in the pseudocolor scale bar (red regions, apical; blue regions, basal). Images are from a representative experiment after 60 minutes of culture. (B) HUVECs monolayers grown as in panel A were treated with either EFNA4–Fc protein complexes (preclustered with biotinylated anti-His Ab followed by streptavidin–Alexa Fluor 405) or cross-linking anti–ICAM-1 mAb (anti–ICAM-1 primary Ab preclustered with anti–mouse Alexa Fluor 488 Ab, green). Cultures were fixed after 15, 30, or 60 minutes and immunostained to analyze the colocalization of clustered EphA2 (anti-EphA2 plus Alexa Fluor 546 secondary Ab) with ICAM-1 (Alexa Fluor 488 Ab) or VCAM-1 (Alexa Fluor 633 Ab) or of EphA2 (Alexa Fluor 546) or VCAM-1 (Alexa Fluor 633 Ab) with ICAM-1, respectively. Z-series confocal images were acquired as in panel A. Fluorescent images are from a representative experiment of EFNA4–Fc treatment. Fluorescent blue-red spots, corresponding to EFNA4-Fc aggregated EphA2 (left), were identified as objects through color thresholding (Metamorph software; white regions). The percentage of these regions containing ICAM-1 (green, right), was measured to determine EphA2 colocalization. The graphs represent the results from the corresponding treatments.
Discussion
In the present study, we demonstrate that expression of EFNA4 in B cells can be linked to the TEM process through a novel interaction with endothelial EphA2. Furthermore, in the case of CLL cells, the high expression of EFNA4 negatively mediates in the TEM, possibly regulating their extravasation potential.
A reduced extravasation capacity of CLL cells compared with that of normal B cells is supported by previous studies. Thus, 51Cr labeling of CLL cells demonstrated that they leave the circulation less rapidly than normal B lymphocytes,11 and [3H]thymidine-labeled CLL cells were shown to survive in the circulation of a CLL patient for many weeks without any evidence of extravasation.10 In line with this, several in vitro studies12,26,27 have shown that the CLL cell has a reduced capacity to adhere to and/or transmigrate through endothelial-cell monolayers compared with normal B cells, whereas an increased adhesion to endothelium is observed in the more advanced stages.8,9,27 A recent study13 has also shown a severely impaired in vitro TEM capacity and in vivo homing of human PB CLL cells to murine peripheral LNs compared with normal B lymphocytes, which has been related to a defective integrin expression by CLL cells. We demonstrate that CLL cases with a low EFNA4 expression show a corresponding greater capacity to adhere to and transmigrate through HUVEC monolayers than those with greater expression, in correlation with disease stage and the presence or absence of lymphadenopathy in these patients, respectively.
In other systems, the magnitude of Eph/EFN signaling is largely dictated by the extent of clusterization, leading to opposing responses such as cell adhesion or repulsion.16,18,28,29 Thus, the greater EFNA4 expression in CLL cells than in normal B cells seems to explain their different responses to similar EphA2 endothelial interactions. Accordingly, the EphA2-Fc treatment of CLL cells diminishes the proportion of cell conjugates established between them and HUVECs in an EFNA4 expression–dependent manner. EFNA4 reverse signaling has been reported to act through repulsive signals in other systems like inhibiting sensory neurite outgrowth within the developing mouse skin,30 further suggesting that this molecule can lead to repulsive signals during cell-cell contacts in the TEM process.
EFNA4 signaling can modulate CLL-cell adhesion to endothelium through regulating other CAMs, as previously reported for other Eph/EFN members in different cell types,16 although the specific pathways of EFNAs reverse signaling are largely unknown. In fact, it down-modulated CD44 expression in CLL cells, a CAM implicated in the normal trafficking of lymphocytes but also in CLL, as well as impairing their adhesion to several integrin ligands, including ECM components like FN, VN, laminin, or endothelial CAMs, like ICAM-1 and VCAM-1, the magnitude of these effects being correlated with the levels of its expression.
Our results show that, in the absence of HUVECs, EFNA4 signaling can inhibit the CCL19-mediated chemotaxis of CLL cells whereas CXCL12 or CXCL13 chemotaxis remained unaffected, supporting a major role in the CCR7-mediated migration. In the chemokine TEM assays, when EFNA4-Fc is added to cultures, the inhibitory effects of reverse signaling are not taking place, thus allowing adhesion but also CCL19-mediated migration. A cooperation between CCL19 and EphA2 forward signaling could also contribute to the increased TEM of CLL cells. In the case of CXCL12 and CXCL13, an increased adhesion caused by the absence of EFNA4 signaling is not sufficient for the TEM of CLL, suggesting that either EphA2 signaling is inhibitory for the TEM mediated by these chemokines and/or that they require a bidirectional EphA2-EFNA4 signaling. Further work will be necessary to clarify these hypotheses.
An essential property of Eph-EFN interaction is that it can result in bidirectional signaling into both the Eph- (forward signaling) and the EFN-expressing cells (reverse signaling).16-19,23,24 In our experimental conditions, the use of homodimeric Eph/EFN-Fc fusion proteins blocks bidirectional signaling while inducing Eph receptor clusterization and signaling.25 This results in an imbalance in the bidirectional EphA2-EFNA4 signaling, similar to what is achieved in other systems when over-expressing Eph/EFN truncated forms lacking intracellular signaling domains,29 but allows the cell- or non–cell-autonomous roles of the interacting Eph/EFN molecules to be analyzed. However, it remains to be determined whether a complete absence of EphA2-EFNA4 bidirectional signaling between lymphocytes and endothelium can be compensated or not by other Eph/EFNs expressed by both cell types, further clarifying the redundant or nonredundant role of these molecules in the studied TEM of lymphocytes.
CD31+ endothelial cells in human LNs mainly expressed EphA2 and rarely other members of the Eph family A such as EphA3, EphA4, and EphA8, which are preferentially expressed by nonendothelial cells scattered throughout the LN parenchyma, supporting the view that EFNA4-EphA2 interactions mediate the TEM of normal B cells. EFNA4-Fc treatment of HUVECs similarly affected CLL and normal B-cell TEM, increasing their adhesion and CCL19-dependent and -independent TEM, suggesting that EphA2 signaling into endothelium promotes TEM. One way of proceeding may be in concert with ICAM-1 and VCAM-1 molecules as the internalization of EphA2-EFNA4-Fc complexes in HUVECs was accompanied by ICAM-1 and VCAM-1 sequestration. Indirect evidence has suggested the involvement of EphA2 in leukocyte extravasation in other systems such as in thrombin-induced in vitro up-regulation of ICAM-1 in endothelial cells31 as well as the known up-regulation of endothelial EphA2 upon inflammatory stimuli.32
In accordance with the role played by Eph receptors in regulating cell shape and movements through reorganizing the cytoskeleton,16-19 we propose that EphA2 could act through connecting the adhesive structure formed by ICAM-1 and VCAM-1 with the endothelial cytoskeleton that forms during TEM. The EphA2-EFNA4 interaction could work through regulating adhesion/detachment at discrete sites of contact between both cell types as in the “invasive” podosomes extended by lymphocytes to palpate the surface of the endothelium.33 Detachment at sites of EphA2-EFNA4 interactions could occur through endocytosis of molecular complexes, a mechanism that has been shown to account for Eph/EFN signaling termination.16,17,28,34 Accordingly, when HUVECs were treated in culture with EFNA4-Fc, EphA2-EFNA4Fc complexes underwent rapid cell internalization. Furthermore, EFNA reverse signals can be modulated by cell-surface shedding of the ligand upon binding to EphA receptors through metalloproteases like Adam10/Kuzbanian, which cleaves EFNA2 from the cell surface.35 By this mechanism, Eph-receptor–bearing structures, such as growth cones, change their response to EFNA molecules from adhesion to repulsion.35 Similarly, EphA2-EFNA4 interactions between endothelium and B cells could be actively formed and terminated to proceed with the TEM process in concert with other mechanisms, including those mediated by chemokines.
In summary, our results support the thesis that TEM of B cells can be mediated by EphA2-EFNA4 interactions and that it is largely dictated by the magnitude of EFNA4 reverse signaling into lymphocytes, leading to an overall repulsion when EFNA4 signaling strength overpasses a threshold, as it is the case in CLL. In contrast, the magnitude of EphA2 signaling into endothelial cells induced upon binding different levels of EFNA4 and its impact on B-cells TEM needs further evaluation (Figure 7).

Summary of a hypothetical model of B cell TEM according to EphA2-EFNA4 interaction and its regulation by EFNA4 signaling strength. Low levels of EFNA4 expression, like those present in most of the normal PB B cells, allow TEM progression to proceed through generating EphA2 forward signals into endothelial cells that connect to the adhesion machinery (at least ICAM-1, VCAM-1 CAMs). By contrast, the greater the levels of EFNA4 expression in B cells, as determined in CLL B cells, led to inhibitory reverse signals that impair TEM.
Summary of a hypothetical model of B cell TEM according to EphA2-EFNA4 interaction and its regulation by EFNA4 signaling strength. Low levels of EFNA4 expression, like those present in most of the normal PB B cells, allow TEM progression to proceed through generating EphA2 forward signals into endothelial cells that connect to the adhesion machinery (at least ICAM-1, VCAM-1 CAMs). By contrast, the greater the levels of EFNA4 expression in B cells, as determined in CLL B cells, led to inhibitory reverse signals that impair TEM.
Although other mechanisms might contribute to the development of clinical lymphadenopathy, such as the rate of cell proliferation/death within the infiltrates36 and, presumably, the levels of lymphocyte egress/retention at the LN, interfering CLL cell extravasation might have therapeutic potential through preventing their migration into LN. Signaling through EFNA4 remarkably blocks TEM in vitro, suggesting that management of this molecule could potentially prevent CLL cell extravasation. Further work is necessary, however, to conclusively address these issues.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
We thank patients for contributing to this research. We also thank Dr P. Cardas (Clinica Moncloa, Madrid) for anatomopahologic analysis of reactive LNs, Dr G. Sanz (H.G.U. Clínico San Carlos) for advice with CLL LN, Juan Ayuga (Blood Bank of H.G.U. G. Marañón, Madrid) for critical suggestions, Drs. Juan J. Muñoz and Angeles Vicente for critical suggestions and review of this manuscript, and Alfonso Cortés, Amalia Vázquez, and Carmen Hernández for their support with confocal microscopy and flow cytometry.
This work has been supported by grants from the Spanish Ministry of Health (FIS-PI50571; FIS PI080093) and the Spanish Ministry of Science (BFU 2004-03 132). E.T. was supported by the Spanish Ministry of Health (grants FIS-PIO50571 and FIS RD06/0010/0003).
Authorship
Contribution: L.M.A.-C. designed experiments; A.G.Z. contributed to the experimental design; E.M.T. and L.M.A.-C. performed the experiments; M.B. provided clinical cases, diagnosed CLL, and studied patients' characteristics; J.Z. provided lymph node biopsies and clinical data from these patients; E.M.T. and L.M.A.-C. analyzed data; E.M.T., M.B., A.G.Z., and L.M.A.-C. discussed data; E.M.T. contributed to writing the paper; L.M.A.-C. wrote the paper; M.B. and J.Z. contributed to the paper revision; and A.G.Z. revised the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Luis Miguel Alonso-Colmenar, Microscopy and Cytometry Research Centre, Faculty of Chemistry, Complutense University of Madrid, Ave Ciudad Universitaria, S/N, 28040 Madrid, Spain; e-mail: lmalonso@bio.ucm.es.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal