

Abstract
Abstract 481
Childhood leukemia frequently originates prenatally. Only a small percentage (<1%) of children with recurrent leukemia associated aberrations detected at birth suffer leukemia later on. In addition, no option to treat the preleukemic clone is availabel. Therefore, neither general screening at birth is useful nor preemptive treatment is possible.
The high incidence (5 to 10%) of the transient leukemia (TL) in newborns with trisomy 21 and the high risk to develop a myeloid leukemia of Down Syndrome (ML-DS) within the first 4 years of life supported the hypothesis that the elimination of the preleukemic GATA1 positive clone might prevent leukemia. Prerequisites are the high sensitivity of TL-blasts to cytarabine, the recurrence of the same GATA1-mutated clone and the feasibility to monitor the preleukemic clone.
Since 4/2007 69 children with TL were enrolled the study “Prevention of Myeloid Leukemia in Children with Down Syndrome and Transient Leukemia” (EudraCT 2006-002962-20) ; Germany n=50, The Netherlands n=16, Slovakia n=1, Czech Republic n=2).
Inclusion criteria were met by 52 children (study patients), 17 children were observed only (protocol patients). Table 1 summarizes the patients' characteristics. The TL and ML-DS specific mutations of the transcriptions factor GATA1 have been detected in 60 children (87%), failure of detection were caused by low percentage of blasts (<2%) combined with late diagnosis (≥20 days after birth). The median follow-up within the study group was 1 year (0.2 to 2.3 years).
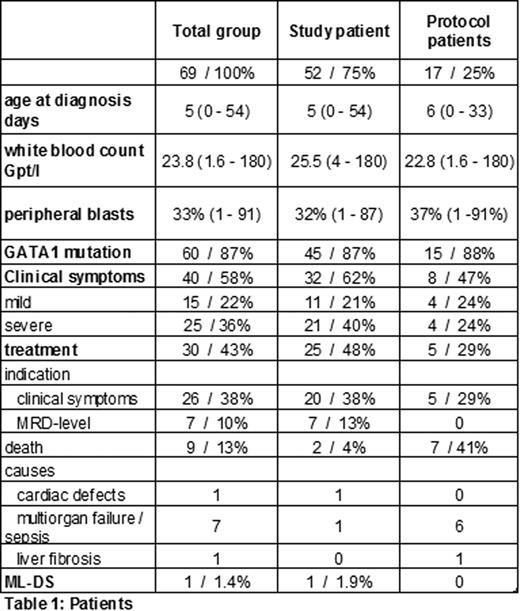
Totally, 58 % of the children showed clinical symptoms associated to the TL, severe complications have been reported in 22 children (table 2). According to the study guidelines 20 out of these 22 children were treated with low dose cytarabine (1.5mg/kg body weight 1 week).
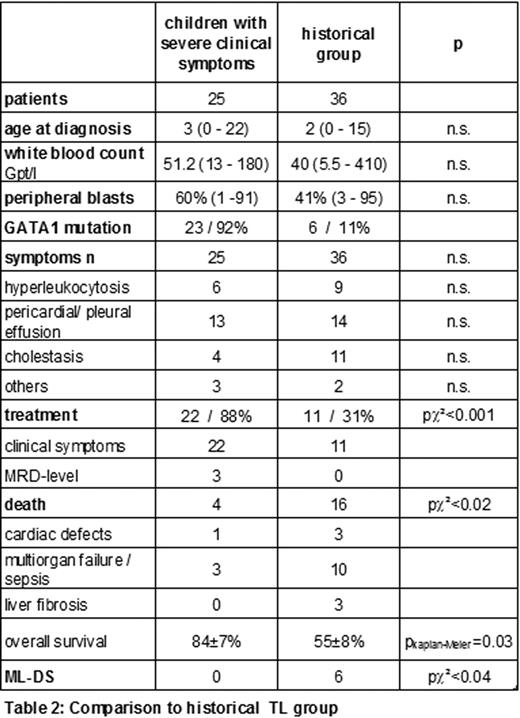
Enrollment to the study including reference diagnostics and consulting, and a consequent treatment seems to improve the prognosis of this particular group. Compared to the historical group of children with similar characteristics (Klusmann et al. Blood 111(6):2991-8, 2008), the overall survival (2 years) significantly increased from 55±7% to 84±8%, p=0.03.
MRD diagnostics by qRT-PCR and/or immunophenotyping was performed in 53 children (77%). Reasons for failure were early deaths (n=9; cardiac defects n=1, prematurity/MOV n=7, liver fibrosis n=1), refusal of monitoring by the parents (n=3), lack of material (n=4). If the MRD-level at week 8 and/or 10 exceeded 10-3 (qRT-PCR) or 10-2 (immunophenotyping), respectively, intervention with low-cytarabine was recommended. Currently, 39 children were already analyzed at week 12 (1st endpoint).
In 7 children (13%) treatment recommendation according to high MRD levels were given. With exception of transient myelosuppression (CTC Grade II) no severe side effects occurred. All children but two became MRD negative at week 12. To date one child with persistent detectable MRD levels suffered ML-DS (1 year after TL).
In summary, participating in the study and treatment of children with TL causing severe clinical symptoms seems to improve the prognosis.
Although the recruitment into the study is faster than expected and the results to date are promising, the follow-up is much too short to draw definitive conclusion.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal