

Abstract
Thrombophilia screening is controversial. In a retrospective family cohort, where probands had thrombosis and a thrombophilic defect, 2479 relatives were tested for thrombophilia. In antithrombin-, protein C–, and protein S–deficient relatives, annual incidences of venous thrombosis were 1.77% (95% CI, 1.14-2.60), 1.52% (95% CI, 1.06-2.11), and 1.90% (95% CI, 1.32-2.64), respectively, at a median age of 29 years and a positive family history of more than 20% symptomatic relatives. In relatives with factor V (FV) Leiden, prothrombin 20210G>A, or high FVIII levels, these were 0.49% (95% CI, 0.39-0.60), 0.34% (95% CI, 0.22-0.49), and 0.49% (95% CI, 0.41-0.51), respectively. High FIX, FXI, and TAFI, and hyperhomocysteinemia were not independent risk factors. Annual incidence of major bleeding in antithrombin-, protein C–, or protein S–deficient relatives on anticoagulants was 0.29% (95% CI, 0.03-1.04). Cumulative recurrence rates in relatives with antithrombin, protein C, or protein S deficiency were 19% at 2 years, 40% at 5 years, and 55% at 10 years. In relatives with FV Leiden, prothrombin 20210G>A, or high levels FVIII, these were 7%, 11%, and 25%, respectively. Considering its clinical implications, thrombophilia testing should address hereditary deficiencies of antithrombin, protein C, and protein S in patients with first venous thrombosis at young age and/or a strong family history of venous thrombosis.
Introduction
Since 1965, an increasing number of coagulation disorders have been identified as risk factors for venous thrombosis. These thrombophilic defects include hereditary deficiencies of antithrombin, protein C and protein S, factor (F) V Leiden, prothrombin 20210G>A, high levels of FVIII, FIX, FXI, and thrombin activatable fibrinolysis inhibitor (TAFI).1-8 In addition, hyperhomocysteinemia showed to be a metabolic thrombophilic defect.9 Together, their prevalence is approximately 25% in the normal population and more than 60% in subjects with venous thrombosis.10 Venous thrombosis is now considered a multicausal disease.10 Gene-gene interactions and environmental risk factors increase the risk of venous thrombosis. Whether patients with venous thrombosis should be tested for thrombophilic defects is still a matter of debate. Previous studies addressed mainly the relative risk of thrombosis for single thrombophilic defects.2-9 However, clinical implications of thrombophilic defects depend on the absolute risk of first venous thrombosis and recurrence rather than the relative risk. Moreover, the absolute risk varies for different thrombophilic defects and may be increased by concomitance of other defects.1,11 To compare the absolute risk for single and combined thrombophilic defects, sufficient numbers of subjects particularly with rare defects are required
We performed a retrospective study in a large series of families to assess the absolute risk of first venous thrombosis and recurrence for currently known thrombophilic defects, either as single or combined defects. We also took into account whether events were idiopathic or provoked.
Methods
Data retrieval
We pooled data of individual subjects from 5 large retrospective family cohort studies with various thrombophilic index defects, which have previously been described (Table 1).1-3,5,12-14 These studies were performed by 3 university hospitals in The Netherlands (University Medical Center Groningen; Academic Medical Center, Amsterdam; Academic Hospital Maastricht; Table 1). Approval was obtained from the institutional review boards of the 3 hospitals. The first study was a single-center study, only performed in Groningen, The Netherlands. It contained first-degree relatives (ie, offspring, siblings, and/or parents) of consecutive patients (probands) with documented venous thrombosis and established hereditary deficiencies of either antithrombin, protein C, or protein S.1,12 As the number of antithrombin-deficient probands was small, second-degree relatives (ie, grandparents and/or blood-related uncles or aunts) with a deficient parent were also identified. Probands were recruited over a 12-year period, and relatives were enrolled between April 1999 and July 2004. Three studies were multicenter studies, performed in Groningen, Amsterdam, and Maastricht, The Netherlands. The study contained first-degree relatives of consecutive patients with venous thrombosis or premature atherosclerosis (< 50 years of age) and the presence of either the prothrombin 20210G>A mutation, high levels of FVIII at repeated measurements, or hyperhomocysteinemia.3,5,13 Enrollment started in May 1998 and was completed in July 2004. The fifth study was a multicenter study (Groningen, Amsterdam, and Maastricht, The Netherlands) of first-degree relatives of consecutive patients with venous thrombosis and FV Leiden who were enrolled between May 1995 and July 1998.2,14
Data retrieval of studies
| Original study | Center | Inclusion period | Probands | Relatives | Reference no. | ||
|---|---|---|---|---|---|---|---|
| Groningen | Amsterdam | Maastricht | |||||
| Antithrombin/protein C/protein S deficiency | Yes | No | No | April 1999–July 2004 | Consecutive patients with venous thrombosis | First-degree relatives, and second degree relatives of antithrombin deficient probands as well | 1,12 |
| Prothrombin 20210G>A | Yes | Yes | Yes | May 1998–July 2004 | Consecutive patients with venous thrombosis, or premature atherosclerosis | First-degree relatives | 3 |
| High factor VIII | Yes | Yes | Yes | May 1998–July 2004 | First-degree relatives | 5 | |
| Hyperhomocysteinemia | Yes | Yes | Yes | May 1998–July 2004 | First-degree relatives | 13 | |
| Factor V Leiden | Yes | Yes | Yes | May 1995–July 1998 | Consecutive patients with venous thrombosis | First-degree relatives | 2,14 |
| Original study | Center | Inclusion period | Probands | Relatives | Reference no. | ||
|---|---|---|---|---|---|---|---|
| Groningen | Amsterdam | Maastricht | |||||
| Antithrombin/protein C/protein S deficiency | Yes | No | No | April 1999–July 2004 | Consecutive patients with venous thrombosis | First-degree relatives, and second degree relatives of antithrombin deficient probands as well | 1,12 |
| Prothrombin 20210G>A | Yes | Yes | Yes | May 1998–July 2004 | Consecutive patients with venous thrombosis, or premature atherosclerosis | First-degree relatives | 3 |
| High factor VIII | Yes | Yes | Yes | May 1998–July 2004 | First-degree relatives | 5 | |
| Hyperhomocysteinemia | Yes | Yes | Yes | May 1998–July 2004 | First-degree relatives | 13 | |
| Factor V Leiden | Yes | Yes | Yes | May 1995–July 1998 | Consecutive patients with venous thrombosis | First-degree relatives | 2,14 |
Probands were tested on multiple thrombophilic defects in all 5 studies, including antithrombin, protein C, or protein S deficiency, factor V Leiden, prothrombin 20210G>A, and high factor VIII levels (Table 2). When multiple defects were demonstrated, probands were randomly assigned to one of the simultaneously performed studies.1,3,5,12,13 In the present study, probands were classified in the following order of index defects: antithrombin deficiency, protein C deficiency, protein S deficiency, FV Leiden, prothrombin 20210G>A, high factor VIII levels, and hyperhomocysteinemia. Primary thrombophilic (index) defect status was reassessed in the FV Leiden study using the same strategy (eg, when a proband included in the FV Leiden study also was protein S deficient, this proband was reassigned to a protein S–deficient family in the pooled study). This order was based on the conception that of (genetic) thrombophilic defects, antithrombin deficiency is associated with the highest risk of first venous thrombosis and hyperhomocysteinemia, with the lowest risk.13,15 Furthermore, inclusion was not yet stopped at the time when articles on some of the original studies were written.2,3,5 Therefore, included numbers from the pooled study and original studies can differ.
Data retrieval of probands and their enrolled relatives
| Original study | Pooled study | |||||||
|---|---|---|---|---|---|---|---|---|
| Antithrombin deficient, no. | Protein C deficient, no. | Protein S deficient, no. | FV Leiden, no. | Prothrombin 20210G>A, no. | High FVIII, no. | Hyperhomocysteinemia, no. | Total, no. | |
| Antithrombin/protein C/protein S deficiency, probands, no. | 12 | 40 | 72 | 0 | 0 | 0 | 0 | 124 |
| Prothrombin 20210G>A/high factor VIII/hyperhomocysteinemia, probands, no. | 0 | 2 | 1 | 69 | 138 | 173 | 163 | 546 |
| Factor V Leiden, probands, no. | 0 | 4 | 1 | 202 | 0 | 0 | 0 | 207 |
| Total probands, no. | 12 | 46 | 74 | 271 | 138 | 173 | 163 | 877 |
| Relatives enrolled, no. | 97 | 154 | 332* | 557 | 346 | 576 | 417 | 2479 |
| Original study | Pooled study | |||||||
|---|---|---|---|---|---|---|---|---|
| Antithrombin deficient, no. | Protein C deficient, no. | Protein S deficient, no. | FV Leiden, no. | Prothrombin 20210G>A, no. | High FVIII, no. | Hyperhomocysteinemia, no. | Total, no. | |
| Antithrombin/protein C/protein S deficiency, probands, no. | 12 | 40 | 72 | 0 | 0 | 0 | 0 | 124 |
| Prothrombin 20210G>A/high factor VIII/hyperhomocysteinemia, probands, no. | 0 | 2 | 1 | 69 | 138 | 173 | 163 | 546 |
| Factor V Leiden, probands, no. | 0 | 4 | 1 | 202 | 0 | 0 | 0 | 207 |
| Total probands, no. | 12 | 46 | 74 | 271 | 138 | 173 | 163 | 877 |
| Relatives enrolled, no. | 97 | 154 | 332* | 557 | 346 | 576 | 417 | 2479 |
Probands were classified according to their index defect. In case of multiple defects, the index defect was chosen in the following order: antithrombin deficiency, protein C deficiency, protein S deficiency, FV Leiden, prothrombin 20210G>A, high factor VIII levels, hyperhomocysteinemia. For example, a proband with protein C deficiency and high factor VIII levels, and his first-degree relatives were classified as a protein C–deficient family and not as a family with high factor VIII levels.
One-hundred forty-nine protein S type I–deficient relatives, 183 protein S type III–deficient relatives. Protein S type III deficiency was not considered a risk factor in further analyses.
Subjects
All relatives, identified by pedigree analysis, were 15 years or older and were contacted through the probands. All participants provided written informed consent in accordance with the Declaration of Helsinki. Physicians at the thrombosis outpatient clinics collected detailed information about previous episodes of venous thrombosis, exposure to exogenous risk factors for venous thrombosis, and anticoagulant treatment using a validated questionnaire,16 and by reviewing medical records. Clinical data were collected prior to laboratory testing. Relatives were tested for hereditary deficiencies of antithrombin, protein C and protein S, FV Leiden, prothrombin 20210G>A, and high levels of FVIII, regardless their index defects. In addition, high levels of FIX, FXI, and TAFI, and hyperhomocysteinemia were measured in most relatives as well, but not all due to shortage of stored plasma or the inability to perform homocysteine tests for which relatives were asked to return to our outpatient clinic in a fasting state.
Laboratory studies
Thrombophilic defects were identified using the same assays in all 3 hospitals and in all 5 studies. Activity of antithrombin (Chromogenix, Mölndal, Sweden) and protein C (Behring, Marburg, Germany) was measured by chromogenic substrate assays, and protein C and protein S antigen levels by enzyme-linked immunosorbent assay (ELISA; DAKO, Glostrup, Denmark). Normal ranges were determined in healthy volunteers who had no family history of venous thrombosis, were not pregnant, and had not used oral contraceptives for at least 3 months. Antithrombin deficiency was defined by decreased levels of antithrombin activity (ie, below the lower limit of its normal range: < 70 IU/dL). Protein S deficiency type I was defined by decreased free and total protein S levels (both < 65 IU/dL).17 Protein S deficiency type III was defined by decreased free and normal total protein S levels. Protein S type III deficiency was separately analyzed in a previous article of ours, where it showed to be an independent risk factor for venous thrombosis when free protein S levels were much lower than the lower limit of the normal range.18 Protein C deficiency type I and type II were defined by decreased levels or activity of protein C antigen (both < 65 IU/dL). Deficiencies were considered inherited if they were confirmed by measuring a second sample that was collected 3 months later and were found in at least 2 family members. If there was a discrepancy between the results of the 2 tests, a third sample was tested. A deficiency was considered acquired, through use of oral contraceptives or pregnancy, unless it was confirmed at least 3 months after withdrawal of oral contraceptives or delivery, respectively. FV Leiden and prothrombin 20210G>A were demonstrated by polymerase chain reactions.19,20 Factors VIII:C, IX:C, and XI:C were measured by one-stage clotting assays and were increased at levels above 150 IU/dL, to enable a comparison of results between our and other studies.4-7 TAFI activity was measured in one laboratory by chromogenic substrate assay (Pefakit; Pentafarm, Basel, Switzerland). TAFI levels above the 95th percentile were defined as increased (> 125 U/dL). Levels of homocysteine were measured by high-performance liquid chromatography after overnight fasting.21 Hyperhomocysteinemia was defined as a fasting homocysteine level higher than 18.5 μM.9 Lupus anticoagulant and anticardiolipin antibodies, using previously described tests,1 were rarely demonstrated and therefore not evaluated. If a relative had prevalent venous thrombosis at the time of inclusion (ie, end of follow-up), thrombophilia testing was postponed and performed 3 months after onset of the event. No relatives were tested for thrombophilia when they had metastasized malignant disease. If relatives were on treatment with acenocoumarol, a short-acting vitamin K antagonist, blood samples were taken after treatment had been interrupted for at least 2 weeks; meanwhile, nadroparin was given subcutaneously. Additional tests were performed on plasma stored at − 80°C from relatives who had been enrolled at a time when 1 or more of these deficiencies or defects had not yet been recognized as risk factors for venous thrombosis.
Adjudication of clinical end points
Venous thrombosis was considered established if deep vein thrombosis was confirmed by compression ultrasound or venography, and pulmonary embolism by ventilation and perfusion lung scanning, spiral CT scanning, or pulmonary angiography, or when the patient had received full-dose heparin and a vitamin K antagonist for at least 3 months without objective testing at a time when these techniques were not yet available. Provoked venous thrombosis was defined if it had occurred at or within 3 months after exposure to exogenous risk factors including surgery, trauma, immobilization for more than 7 days, pregnancy, puerperium, the use of oral contraceptives or hormonal replacement therapy, or malignancy. In the absence of these risk factors, venous thrombosis was considered idiopathic.
From relatives with an antithrombin, protein C, or protein S deficiency, we retrieved information on bleeding events associated with the treatment with vitamin K antagonists. Clinically overt bleedings, which required hospitalization or blood transfusion, were intracranial or retroperitoneal, or if they led directly to death were classified as major.
Statistical analysis
We assessed the absolute risk of venous thrombosis (ie, provoked or idiopathic) in relatives with deficiencies of antithrombin, protein C, or protein S, FV Leiden, prothrombin 20210G>A, or high levels of FVIII. Relatives with more than one thrombophilic defect were assigned to each of the corresponding subgroups, to calculate the absolute risk for each defect separately, either as single defect or as combined defects. Relatives were excluded when the laboratory set of these 6 thrombophilic defects was not completed. Probands were excluded from analysis to avoid bias. Possible interactions between high FVIII levels and high levels of FIX, FXI, or TAFI, or hyperhomocysteinemia were analyzed by comparing the risk of venous thrombosis for each of the latter defects separately and in combination with high FVIII levels to the risk in relatives without these defects. Observation time was defined as the period from the age of 15 years until the first thrombotic episode or the end of study. Clinical data and blood samples were collected at the end of the observation period. Hence, treatment and clinical outcome were not influenced by the results of thrombophilia testing.
Event-free survival for first venous thrombosis and recurrence, respectively, were analyzed by the Kaplan-Meier method. Cumulative recurrence rates were calculated over the period from the end of anticoagulant treatment after the first episode of venous thrombosis until either the date of first recurrence or the end of study. We pooled data when cumulative incidences of first venous thrombosis in relatives with different thrombophilic index defects were comparable, to assess the effects of exogenous risk factors. Relative risks were adjusted for age and sex using a Cox regression model with venous thrombosis as dependent variable and thrombophilic defects as independent variables, including interaction terms. To account for the nonrandomness of the relatives analyzed, outcome rates were also adjusted for clustering within families. Therefore, all relatives were identified by a family number, which included a code for the type of thrombophilic defect. Adjustment for clustering was performed with Cox regression analysis and the robust sandwich method (in SAS 9.1; SAS Institute, Cary, NC), resulting in adjusted relative risks with 95% confidence limits and P values.
Prevalence of venous thrombosis in relatives of probands with a certain thrombophilic defect was calculated by dividing the number of symptomatic relatives of probands with thrombophilic defect “x” by total number of their relatives.
Continuous variables are expressed as median values and ranges; categoric data, as counts and percentages. Differences between groups were evaluated by Student t test or Mann-Whitney U test, depending on the normality of data for continuous data, and by Fisher exact test for categoric data. A 2-tailed P value less than .05 indicates statistical significance. The 95% confidence intervals (95% CI) around the incidence rates were calculated under the Poisson distribution assumption.
Statistical analyses were performed using SAS software, version 9.1.
Results
Clinical characteristics
Our study cohort contained 877 probands with 5202 relatives, who were 15 years or older (Figure 1). Of probands with venous thrombosis (n = 746), median age at enrollment was 46 years (range, 9-89 years), and median age at onset of first venous thrombosis was 34 years (range, 8-88 years). Median age at onset of first venous thrombosis was 28 years (range, 12-68 years) in probands with antithrombin, protein C, or protein S deficiency and 35 years (range, 8-88 years) in probands with FV Leiden, prothrombin 20210G>A, or elevated FVIII levels. Prevalence of venous thrombosis in relatives of probands with antithrombin, protein C, or protein S deficiency was 83 (21%) of 400, which was higher than in relatives of probands with FV Leiden, prothrombin 20210G>A, or high factor VIII levels (122/1479; 8%, P < .001) or in relatives of probands with none of these thrombophilic abnormalities (24/600; 4%, P < .001). To avoid selection bias, probands were not further analyzed. Of relatives, 1057 (20%) had died before the start of the study, including 21% of relatives of probands with deficiencies of antithrombin, protein C, or protein S, and 20% of relatives of probands with other defects. Another 1412 relatives did not participate because of various reasons (exclusion rate, 34%), including refusal or inability to give informed consent or residence outside The Netherlands, and 254 relatives were not evaluable because the set of thrombophilic tests was not performed completely. Forty-five percent were male. Median age at enrollment was 46 years (range, 15-92 years). Median observation period was 30 years (range, 0-77 years). Venous thrombosis had occurred in 229 relatives (9%). Of these events, 133 (58%) were diagnosed with objective techniques; 91 (40%) were idiopathic, 32 (14%) were associated with oral contraceptives (in women, 22%), 40 (17%) were associated with pregnancy/puerperium (in women, 29%), 65 (28%) were associated with surgery, trauma, or immobilization; and 1 (0.4%) was associated with malignancy. Median age at onset of the first episode of venous thrombosis was 35 years (range, 15-84 years). Relatives with antithrombin, protein C, or protein S deficiency had venous thrombosis at younger age than relatives with FV Leiden, prothrombin 20210G>A, or high FVIII levels (median, 29 years vs 40 years; P < .001), either idiopathic (median, 34 years vs 53 years; P < .001) or provoked (median, 27 vs 34 years; P = .022).

Flow diagram of the family cohort. * indicates probands were classified according to their index defect. In case of multiple defects, the index defect was chosen in the following order: antithrombin deficiency, protein C deficiency, protein S deficiency, FV Leiden, prothrombin 20210G>A, high factor VIII levels, and hyperhomocysteinemia.
Flow diagram of the family cohort. * indicates probands were classified according to their index defect. In case of multiple defects, the index defect was chosen in the following order: antithrombin deficiency, protein C deficiency, protein S deficiency, FV Leiden, prothrombin 20210G>A, high factor VIII levels, and hyperhomocysteinemia.
Absolute risks of first venous thrombosis
Annual incidences of venous thrombosis in relatives with antithrombin, protein C, or protein S deficiency, possible concomitance of other thrombophilic defects not taken into account, were 1.77% (95% CI, 1.14-2.60), 1.52% (95% CI, 1.06-2.11), and 1.90% (95% CI, 1.32-1.64), respectively, and in relatives with FV Leiden, prothrombin 20210G>A, or high FVIII levels were 0.49% (95% CI, 0.39-0.60), 0.34% (95% CI, 0.22-0.49), and 0.49% (95% CI, 0.41-0.51), respectively (Table 1). A total of 951 relatives had no thrombophilic defects (27 028 observation years), of which 12 had a first VTE; annual incidence was 0.05% (95% CI, 0.02-0.08). Including the 254 relatives who were not completely tested for these 6 thrombophilic defects did not substantially change risk estimates. Survival analysis showed that relatives with these defects were at continuous risk of venous thrombosis compared with relatives without thrombophilic defects (P < .001; Figure 2A). Because annual and cumulative incidences of first venous thrombosis were similar in antithrombin-, protein C–, or protein S–deficient relatives, as were these in relatives with FV Leiden, prothrombin 20210G>A, or high FVIII levels, relatives were pooled in 2 groups to compare provoked versus idiopathic venous thrombosis (Figure 2B). Provoked events were observed more frequently at age 20 to 35 years. This difference was more pronounced in the group of relatives with anticoagulant deficiencies. Overall, lifetime risk of first venous thrombosis was comparable whether the first episode was provoked or not.

Event-free survival of first venous thrombosis in relatives with thrombophilic defects. * indicates that when analyzing idiopathic events, provoked events were censored and vice versa.
Event-free survival of first venous thrombosis in relatives with thrombophilic defects. * indicates that when analyzing idiopathic events, provoked events were censored and vice versa.
Interactions of FVIII, and FIX, FXI, TAFI, and hyperhomocysteinemia
Relatives with high levels of FIX, FXI, or TAFI, or hyperhomocysteinemia were at risk of venous thrombosis only when they also had high FVIII levels (Table 4). Adjusted relative risks were 1.5 (95% CI, 0.9-2.3), 2.4 (95% CI, 1.5-3.8), 1.8 (95% CI, 1.0-3.6), and 2.9 (95% CI, 1.6-5.3), respectively, compared with relatives with normal levels. Excluding relatives with high FVIII levels, these were 0.3 (95% CI, 0.1-0.8), 0.8 (95% CI, 0.3-2.1), 0.5 (95% CI, 0.2-1.3), and 0.6 (95% CI, 0.2-1.4), respectively. As the risk of thrombosis associated with these defects was apparently due to high FVIII levels, they were not further considered.
Combined thrombophilic defects
The absolute risks of venous thrombosis for single thrombophilic defects and combinations are summarized in Table 5. Compared with the annual incidence of venous thrombosis in relatives with none of these defects (0.05%; 95% CI, 0.02-0.08), annual incidences were elevated in relatives with single heterozygous prothrombin 20210G>A (0.19%; 95% CI, 0.08-0.38), high FVIII levels (0.23%; 95% CI, 0.16-0.33), and heterozygous FV Leiden (0.22%; 95% CI, 0.13-0.33). In relatives with a single deficiency of protein C, antithrombin, or protein S these were 0.54% (95% CI, 0.15-1.39), 1.15% (95% CI, 0.42-2.50), and 1.45% (95% CI, 0.66-2.75), respectively. Concomitance of one or more other defects increased the absolute risk of venous thrombosis markedly. This effect of concomitance decreased in relatives with FV Leiden, prothrombin 20210G>A, or high FVIII levels when relatives with concomitant deficiencies of antithrombin, protein C, or protein S were excluded. Double heterozygosity of FV Leiden and prothrombin 20210G>A or/and homozygosity of these mutations was demonstrated in 81 relatives: 23 homozygotes of FV Leiden (8 events; annual incidence, 1.30%; 95% CI, 0.56-2.56); 8 homozygotes of prothrombin 20210G>A (no events); 50 double heterozygotes of these mutations (7 events; annual incidence, 0.48%; 95% CI, 0.19-0.99), 6 homozygotes of FV Leiden who also were heterozygotes of prothrombin 20210G>A (1 event); and 3 homozygotes of prothrombin 20210G>A who were also heterozygotes of FV Leiden (no events). Relatives with these genotypes are hereafter indicated as “double heterozygous/homozygous.”
Risk of first venous thrombosis in 2479 relatives of 877 probands associated with thrombophilic defects
| Index defect* | Observation years | Relatives with event | Annual incidence, % (95% CI) | Adjusted relative risk† (95% CI) |
|---|---|---|---|---|
| Antithrombin deficiency, n = 60 | 1416 | 25 | 1.77 (1.14-2.60) | 28.2 (13.5-58.6) |
| Protein C deficiency, n = 91 | 2301 | 35 | 1.52 (1.06-2.11) | 24.1 (13.7-42.4) |
| Protein S deficiency, n = 94 | 1846 | 35 | 1.90 (1.32-2.64) | 30.6 (26.9-55.3) |
| High FVIII, n = 776 | 26 315 | 130 | 0.49 (0.41-0.51) | 7.1 (4.3-11.8) |
| Factor V Leiden, n = 652 | 18 237 | 89 | 0.49 (0.39-0.60) | 7.5 (4.4-12.6) |
| Prothrombin 20210G>A, n = 288 | 8324 | 28 | 0.34 (0.22-0.49) | 5.2 (2.8-9.7) |
| Index defect* | Observation years | Relatives with event | Annual incidence, % (95% CI) | Adjusted relative risk† (95% CI) |
|---|---|---|---|---|
| Antithrombin deficiency, n = 60 | 1416 | 25 | 1.77 (1.14-2.60) | 28.2 (13.5-58.6) |
| Protein C deficiency, n = 91 | 2301 | 35 | 1.52 (1.06-2.11) | 24.1 (13.7-42.4) |
| Protein S deficiency, n = 94 | 1846 | 35 | 1.90 (1.32-2.64) | 30.6 (26.9-55.3) |
| High FVIII, n = 776 | 26 315 | 130 | 0.49 (0.41-0.51) | 7.1 (4.3-11.8) |
| Factor V Leiden, n = 652 | 18 237 | 89 | 0.49 (0.39-0.60) | 7.5 (4.4-12.6) |
| Prothrombin 20210G>A, n = 288 | 8324 | 28 | 0.34 (0.22-0.49) | 5.2 (2.8-9.7) |
All P values were less than .001.
As concomitance of defects occurred frequently, relatives could be counted twice or more.
Adjusted for age, sex, and clustering in families and compared with relatives with no thrombophilic defects (n=951; 27 028 observation years; 12 events; annual incidence 0.05%; 95% CI, 0.02-0.08).
Annual incidences of first episodes of venous thrombosis in relatives with high levels of FIX, FXI, or TAFI, or hyperhomocysteinemia
| Observation years | Relatives with event | Annual incidence, % (95% CI) | Adjusted relative risk* (95% CI) | P | |
|---|---|---|---|---|---|
| High FIX levels | |||||
| Absent, n=1684 | 48 702 | 124 | 0.25 (0.21-0.30)† | Reference | |
| Present, n=280 | 9751 | 24 | 0.25 (0.16-0.37) | 1.0 (0.5-1.5) | .71 |
| With normal FVIII levels, n=136 | 4400 | 3 | 0.07 (0.02-0.20) | 0.3 (0.1-0.8) | .016 |
| With high FVIII levels, n=144 | 5321 | 21 | 0.39 (0.24-0.60) | 1.5 (0.9-2.3) | .11 |
| High FXI levels | |||||
| Absent, n=2192 | 64 350 | 179 | 0.28 (0.24-0.32)‡ | Reference | |
| Present, n=148 | 4798 | 25 | 0.52 (0.33-0.77) | 2.2 (1.3-3.5) | .002 |
| With normal FVIII levels, n=62 | 1858 | 4 | 0.22 (0.06-0.55) | 0.8 (0.3-2.1) | .63 |
| With high FVIII levels, n=86 | 2940 | 21 | 0.71 (0.44-1.09) | 2.4 (1.5-3.8) | < .001 |
| High TAFI levels | |||||
| Absent, n=1824 | 53 044 | 149 | 0.28 (0.24-0.33)§ | Reference | |
| Present, n=203 | 7257 | 21 | 0.29 (0.18-0.44) | 1.0 (0.6-1.6) | .97 |
| With normal FVIII levels, n=121 | 4161 | 6 | 0.14 (0.05-0.31) | 0.5 (0.2-1.3) | .15 |
| With high FVIII levels, n=82 | 3095 | 15 | 0.49 (0.27-0.80) | 1.8 (1.0-3.6) | .073 |
| Hyperhomocysteinemia | |||||
| Absent, n=1642 | 48 075 | 127 | 0.26 (0.22-0.31)¶ | Reference | |
| Present, n=190 | 6507 | 25 | 0.38 (0.25-0.57) | 1.7 (1.0-2.9) | .05 |
| With normal FVIII levels, n=112 | 3380 | 5 | 0.14 (0.05-0.34) | 0.6 (0.2-1.4) | .20 |
| With high FVIII levels, n=78 | 3077 | 20 | 0.65 (0.40-1.00) | 2.9 (1.6-5.3) | < .001 |
| Observation years | Relatives with event | Annual incidence, % (95% CI) | Adjusted relative risk* (95% CI) | P | |
|---|---|---|---|---|---|
| High FIX levels | |||||
| Absent, n=1684 | 48 702 | 124 | 0.25 (0.21-0.30)† | Reference | |
| Present, n=280 | 9751 | 24 | 0.25 (0.16-0.37) | 1.0 (0.5-1.5) | .71 |
| With normal FVIII levels, n=136 | 4400 | 3 | 0.07 (0.02-0.20) | 0.3 (0.1-0.8) | .016 |
| With high FVIII levels, n=144 | 5321 | 21 | 0.39 (0.24-0.60) | 1.5 (0.9-2.3) | .11 |
| High FXI levels | |||||
| Absent, n=2192 | 64 350 | 179 | 0.28 (0.24-0.32)‡ | Reference | |
| Present, n=148 | 4798 | 25 | 0.52 (0.33-0.77) | 2.2 (1.3-3.5) | .002 |
| With normal FVIII levels, n=62 | 1858 | 4 | 0.22 (0.06-0.55) | 0.8 (0.3-2.1) | .63 |
| With high FVIII levels, n=86 | 2940 | 21 | 0.71 (0.44-1.09) | 2.4 (1.5-3.8) | < .001 |
| High TAFI levels | |||||
| Absent, n=1824 | 53 044 | 149 | 0.28 (0.24-0.33)§ | Reference | |
| Present, n=203 | 7257 | 21 | 0.29 (0.18-0.44) | 1.0 (0.6-1.6) | .97 |
| With normal FVIII levels, n=121 | 4161 | 6 | 0.14 (0.05-0.31) | 0.5 (0.2-1.3) | .15 |
| With high FVIII levels, n=82 | 3095 | 15 | 0.49 (0.27-0.80) | 1.8 (1.0-3.6) | .073 |
| Hyperhomocysteinemia | |||||
| Absent, n=1642 | 48 075 | 127 | 0.26 (0.22-0.31)¶ | Reference | |
| Present, n=190 | 6507 | 25 | 0.38 (0.25-0.57) | 1.7 (1.0-2.9) | .05 |
| With normal FVIII levels, n=112 | 3380 | 5 | 0.14 (0.05-0.34) | 0.6 (0.2-1.4) | .20 |
| With high FVIII levels, n=78 | 3077 | 20 | 0.65 (0.40-1.00) | 2.9 (1.6-5.3) | < .001 |
Numbers of relatives tested for levels of FIX, FXI, TAFI, and homocysteine were 1964, 2340, 2027, and 1832, respectively.
Adjusted for age, sex, and clustering of thrombophilic defects in families.
Annual incidence in relatives with normal FIX and FVIII levels was 0.17% (95% CI, 0.13-0.22).
Annual incidence in relatives with normal FXI and FVIII levels was 0.18% (95% CI, 0.15-0.23).
Annual incidence in relatives with normal TAFI and FVIII levels was 0.18% (95% CI, 0.14-0.23).
Annual incidence in relatives with normal homocysteine and FVIII levels was 0.19% (95% CI, 0.15-0.24).
Risk of first venous thrombosis associated with cosegregation
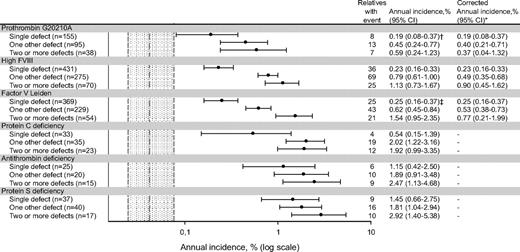
Annual incidence, % (log scale).
Dotted gray area represents annual incidence of venous thrombosis in relatives without any defect (0.05%), with its 95% confidence interval (0.02%-0.08%).
*Excluding relatives with hereditary antithrombin, protein C, or protein S deficiency.
†One-hundred fifty-one single heterozygous prothrombin 20210G>A; annual incidence was 0.19% (95% CI, 0.08-0.38).
‡Three-hundred fifty-nine single heterozygous factor V Leiden; annual incidence was 0.22% (95% CI, 0.13-0.33).
Absolute risks of recurrent venous thrombosis
After their first episode of venous thrombosis, relatives received anticoagulant treatment for a median time of 6 months (range, 3-256 months). At enrollment, 42 relatives still received anticoagulant treatment. None of them experienced a recurrence. Of the remaining 187 relatives, 60 (32%) had antithrombin, protein C, or protein S deficiency, and 110 (59%) had FV Leiden, prothrombin 20210G>A, or high FVIII levels. There were 69 recurrences, of which 36 (52%) were diagnosed with objective techniques. Of antithrombin-, protein C–, or protein S–deficient relatives, 42% received anticoagulant treatment for more than 6 months (median, 14 months), compared with 35% of relatives with FV Leiden, prothrombin 20210G>A, or high FVIII levels (median, 12 months; P = .32). Median time of anticoagulant treatment in relatives with provoked or idiopathic first venous thrombosis was 6 months in both groups, whereas 40% of relatives with provoked first venous thrombosis received anticoagulant treatment for 3 months versus 30% of relatives with idiopathic first venous thrombosis (P = .51). Cumulative recurrence rates in relatives (not on continuing anticoagulant treatment) with antithrombin, protein C, or protein S deficiency were 19% after 2 years, 40% after 5 years, and 55% after 10 years. Median age at recurrence was 36 years (range, 20-75 years); annual incidence was 6.23% (95% CI, 4.31-8.70). Log-rank test revealed no statistical differences whether first event was idiopathic or provoked (52% vs 59%; P = .93). In relatives with FV Leiden, prothrombin 20210G>A, or high FVIII levels, recurrence rates were 7%, 11%, and 25%, respectively, at a median age of 43 years (range, 21-85 years) and an annual incidence of 2.25% (95% CI, 1.52-3.21; Figure 3). Recurrence rates were 29% after an idiopathic first event and 24% after a provoked first event (P = .91). Numbers of double heterozygous/homozygous carriers of FV Leiden or/and prothrombin 20210G>A were too small to obtain accurate estimates of recurrence rates.

Event-free survival of recurrent venous thrombosis in relatives of probands with thrombophilic defects.
Event-free survival of recurrent venous thrombosis in relatives of probands with thrombophilic defects.
Major bleeding
Major bleeding was observed in 2 relatives with antithrombin, protein C, or protein S deficiency while they were treated with vitamin K antagonists, including relatives on long-term treatment; annual incidence was 0.29% (95% CI, 0.03-1.04). One relative was a 38-year-old man with protein C deficiency, the other relative was a 73-year-old man with antithrombin deficiency; both had a hemorrhagic stroke.
Discussion
This study shows that thrombophilic defects can be classified as strong and mild risk factors for venous thrombosis. Strong risk factors included hereditary deficiencies of antithrombin, protein C, and protein S, whereas heterozygous FV Leiden, heterozygous prothrombin 20210G>A, and high FVIII levels were mild risk factors. Although high levels of FIX, FXI, and TAFI, and hyperhomocysteinemia were identified as risk factors for venous thrombosis as reported before,4-9 the associated risks appeared to be due to concomitance of high factor VIII levels. Although these results should be interpreted with some caution due to multiple testing, they are in line with previous studies that reported on this issue.6,8,14,22 When concomitance of other thrombophilic defects was not taken into account, the annual risk of first venous thrombosis in relatives with a strong thrombophilic defect was 1.77% to 1.90% and 0.34% to 0.49% in relatives with a mild thrombophilic defect, which is 15- to 19-fold and 3- to 5-fold higher compared with the community, respectively.23,24
Because combinations of thrombophilic defects were frequently observed (ie, 60% of relatives), we also estimated the absolute risk for single and combined defects. The annual incidence in relatives with a single strong thrombophilic defect ranged from 0.54% to 1.45% and increased to 1.92% to 2.92% when it was combined with 2 or more other defects. In relatives with a single mild defect, annual incidence ranged from 0.19% to 0.25% and increased to 0.59% to 1.54%. Annual incidence was 0.05% in relatives without any of these defects. The higher risk in relatives with a combination of a mild defect and another defect was partly due to concomitance of a strong thrombophilic defect. These results support the concept of multicausality,10,11 but due to the small numbers in subgroups, the results must be seen as illustrative rather than confirmative.
Exogenous risk factors had an additional effect on the risk of first venous thrombosis, particularly in relatives with a strong thrombophilic defect at age 35 years or younger. This effect was due mainly to the use of oral contraceptives and pregnancy/puerperium, which are the most prevalent exogenous risk factors at young age. However, exogenous risk factors did not influence lifetime risk of first venous thrombosis.
Strong and mild thrombophilic defects could also be classified with respect to the risk of recurrent venous thrombosis. Recurrence rates at 5 and 10 years ranged from 40% to 55% and from 11% to 25%, respectively, compared with 22% to 30% in the community.25 This finding is in agreement with previous studies that did not demonstrate an increased risk of recurrence in subjects with mild thrombophilic defects.26,27
Our data show that only strong thrombophilic defects may have clinical implications in patients with venous thrombosis and their relatives. Since the risk of recurrence remained high over at least 5 to 10 years after the first episode of venous thrombosis in relatives with a strong thrombophilic defect, it could be considered to extend this treatment for at least 5 to 10 years. Temporary prophylaxis could be considered in high-risk situations, as the risk of first venous thrombosis is high in these subjects (nearly 2% per year), although the benefits of this strategy are not evidence based. Although the risk of prolonged anticoagulant treatment includes major bleeding, it was only 0.29% per year (95% CI, 0.03-1.04) in these relatives. This risk is lower than we previously reported in unselected patients with venous thrombosis, in whom treatment with vitamin K antagonists was monitored by anticoagulation clinics in The Netherlands (2.8% per year), as were these relatives.28 Perhaps strong thrombophilic defects protect against bleeding during anticoagulant treatment. However, it may also be due to the younger age of these relatives at time of their first thrombotic event. Nevertheless, the low risk of major bleeding will diminish the reluctance to extend anticoagulant treatment. Moreover, none of the relatives with strong thrombophilic defects had a recurrence while on anticoagulant treatment. This is in line with a recent publication on this issue, wherein patients with thrombophilia and venous thrombosis had no increased risk of recurrence compared with patients without thrombophilia while on warfarin therapy.29
It should, however, be noted that antithrombin, protein C, or protein S deficiencies are rare, even in patients with venous thrombosis.10 A strong positive family history of venous thrombosis (ie, > 20% of relatives) could be used in our study to identify these subjects. Therefore, family history is an important issue in clinical practice, provided a sufficient size of families. Young age was another predictor of a strong thrombophilic defect, considering that the median age at onset of first venous thrombosis was 29 years in relatives with these deficiencies, compared with 62 years in the community.24
The risk of first venous thrombosis and recurrence in double heterozygous/homozygous relatives could not be accurately estimated because numbers were too small. Homozygosity of FV Leiden was the strongest risk factor for venous thrombosis in this subgroup. Previous studies on these risk factors contained small numbers as well (n = 17,24 n = 7,26 and n = 20, respectively27,30,31 ). Although these studies consistently reported an increased risk of first venous thrombosis and recurrence in heterozygous/homozygous patients, we think that the results of our study and previous studies are not conclusive.
Our findings are inconsistent with 2 recent prospective studies on this issue.23,28 These studies reported a similar risk of recurrence in patients with a thrombophilic defect, compared with patients without a defect. However, follow up after first venous thrombosis was only 2 years in one study,32 whereas the other study contained only 25 patients with deficiencies of antithrombin, protein C, or protein S.26 Moreover, the thrombotic potency of different thrombophilic defects was quantified as equal. This approach is not appropriate to assess the associated absolute risk of recurrent venous thrombosis, as we showed that risk estimates are not the same for various thrombophilic defects. A comparison with our findings is hampered because age at onset of first venous thrombosis in these studies (mean, 51 years23 and 67 years, respectively26,32 ), was markedly higher than in our study (median, 35 years). This difference emphasizes that strong thrombophilic defects are risk factors at young age. It also explains why only 1% of first episodes of venous thrombosis in our study were associated with malignancy, compared with 4% to 20% in the normal population.23,33
Some methodological aspects of our study warrant comment. The results may have been influenced by its design of a family cohort study. However, population studies are not suitable for risk assessments in subjects with strong, but rare, thrombophilic defects.26,32 Second, unknown inheritable thrombophilic defects may have influenced our results. This is not likely, as the annual incidence of venous thrombosis in relatives without any of tested thrombophilic defects was only 0.05%, and relative risks were adjusted for clustering within families. Third, events were not always confirmed by objective techniques, because these techniques were not available at the time of event. Therefore, incidences of venous thrombosis may have been overestimated. Since these were compared with incidences from population studies that used the same classification of venous thrombosis,23-25 relative risk estimates would not change. Moreover, we assume that misclassification of venous thrombosis would occur equally in relatives with a thrombophilic defect, and those without a thrombophilic defect, and therefore would not change our relative risk estimates. Fourth, referral bias may have been introduced by the university hospital setting, but was probably reduced by testing consecutive patients with thrombosis. Finally, our exclusion rate may have overstated the risk of thrombosis, as it is conceivable that relatives without thrombosis were less inclined to provide informed consent. On the other hand, an excess of fatal events in relatives with a strong thrombophilic defect might have underestimated the risk of venous thrombosis. However, these defects were not associated with a reduced life expectancy in previous studies,34,35 whereas our study did not show a difference in death rate between relatives of probands with a strong thrombophilic defect and relatives of probands with another defect.
In conclusion, of thrombophilic defects, hereditary deficiencies of antithrombin, protein C, and protein S are associated with an excessively high absolute risk of first and recurrent venous thrombosis. A strong positive family history of venous thrombosis and/or young age could be used in our study to identify these rare subjects. Considering its possible clinical implications, thrombophilia testing should primarily address deficiencies in these patients with first venous thrombosis.
An Inside Blood analysis of this article appears at the front of this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This study was funded by grant 28-2783 from the Prevention Fund/ZonMW (The Hague, The Netherlands) and grant 99.187 from the Dutch Heart Foundation (The Hague, The Netherlands).
The funding organizations are public institutions and had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; and preparation, review, or approval of the paper.
Authorship
Contribution: W.M.L. had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis; S.M., K.H., M.H.P., H.R.B., and J.M. were responsible for study concept and design; M.C., I.B., and J.-L.P.B. acquired the data; W.M.L., N.J.G.M.V., M.H.P., and J.M. analyzed and interpreted the data; W.M.L. drafted the paper; W.M.L., M.C., N.J.G.M.V., I.B., J.-L.P.B., S.M., K.H., M.H.P., and J.M. critically revised the paper for important intellectual content; W.M.L., N.J.G.M., and M.H.P. performed statistical analysis; S.M. and H.R.B. obtained funding; W.M.L., M.C., I.B., and J.-L.P.B. provided administrative, technical, or material support; and S.M., K.H., M.H.P., H.R.B., and J.M. supervised the study.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Willem Lijfering, Division of Haemostasis, Thrombosis and Rheology, Department of Hematology, University Medical Center Groningen, Hanzeplein 1, 9713 GZ Groningen, The Netherlands; e-mail: w.lijfering@int.umcg.nl.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal