

T-cell prolymphocytic leukemia (T-PLL) is consistently associated with inactivation of the ATM gene and chromosomal re-arrangements leading to an overexpression of MTCP1/TCL1 oncoproteins. These alterations are present at the earliest stage of malignant transformation, suggesting that additional events are required for overt malignancy. In this study, we pursued the investigation of the 12p13 deletion, previously shown to occur in approximately half of T-PLLs. We refined the minimal region of deletion by single nucleotide and microsatellite polymorphism allelotyping. We defined a 216-kb region containing the CDKN1B gene that encodes the cyclin-dependent kinase inhibitory protein p27KIP1. Sequencing this gene in 47 T-PLL patient samples revealed a nonsense mutation in one case without 12p13 deletion. The absence of biallelic inactivation of CDKN1B for most patients suggested a haploinsufficiency mechanism for tumor suppression, which was investigated in an animal model of the disease. In a Cdkn1b+/− background, MTCP1 transgenics had consistent and multiple emergences of preleukemic clones not observed in control cohorts. The second Cdkn1b allele was maintained and expressed in these preleukemic clones. Altogether, these data strongly implicate CDKN1B haploinsufficiency in the pathogenesis of T-PLL.
Introduction
T-cell prolymphocytic leukemia (T-PLL) is a rare hematologic disease of elderly people characterized by a mature T-cell phenotype, a large tumoral mass, and an aggressive clinical course. Two genetic alterations are known to play a major role in this disease. First, ATM is somatically deleted or mutated in almost 95% of T-PLL patients (for review, see Stankovic et al1 ), and germinal mutations of ATM responsible for the ataxia telangiectasia (AT) predispose to T-PLL (for review, see Taylor et al2 ). Second, chromosomal translocations and inversions involving TCR genes and MTCP1/TCL1 gene family members are consistently and specifically associated with T-PLL, leading to overexpression of these oncogenes.3,4 The oncogenic role of the MTCP1/TCL1 genes was demonstrated using transgenic mouse models.5,6 Recently, we showed that MTCP1/TCL1 oncogene expression modified T-cell homeostasis,7 which may be related to the impairment of activation-induced cell death (AICD) by these oncoproteins.8 ATM inactivation and MTCP1/TCL1 overexpression are present at the earliest stage of the malignant transformation in clonal T-cell populations frequently found in AT patients. However, the indolent course of these populations suggests that these events are insufficient to induce a leukemic phenotype.9
In the search for additional events crucial for full-blown transformation, we undertook a comparative genomic hybridization analysis in a series of T-PLL cases that revealed numerous recurrent copy number alterations.10 One of these alterations was the deletion of the 12p13 region, which was further characterized by allelotyping. This deletion was shown to occur in almost half of T-PLL cases and the minimal region of deletion (MRD) excluded TEL/ETV6 as a candidate gene.11 However, since this first report, no gene fulfilling classical Knudson criteria for tumor suppression (“2-hit” model) was identified in this region.
Haploinsufficiency has emerged as an important oncogenic event.12 One of the first and best-supported examples is CDKN1B haploinsufficiency. CDKN1B encodes the p27KIP1 protein, which has complex functions in cell-cycle regulation. At high levels, p27KIP1 binds to, and inhibits, cyclinE-CDK2 complexes allowing cell-cycle arrest. Conversely, at lower levels, p27KIP1 stabilizes cyclinD-CDK4/6 complexes, facilitating cell-cycle progression.13 Many reports of hemizygous deletions in various human malignancies, such as lymphoma, acute myeloid leukemia, acute lymphoblastic leukemia, ovarian cancer, and prostate cancer, implicate CDKN1B haploinsufficiency.14,,–17 Underexpression of p27KIP1 has also been identified as a poor prognostic factor in epithelial cancers.18 Furthermore, animal models provide direct evidence for the role of haploinsufficiency in cancer by showing that Cdkn1b hemizygote mice are more sensitive than wild-type animals to mutagenesis, and that tumors arising in this context maintained a Cdkn1b haploinsufficient status.19
Interestingly, CDKN1B locus was located within the 12p13 minimal region of deletion previously defined. By further characterizing this deletion in a series of human T-PLL cases, we showed that CDKN1B is a good candidate to be the target gene of the 12p13 deletion. Then, by modeling allelic loss of Cdkn1b, we showed that p27KIP1 haploinsufficiency contributes to malignant transformation in an animal model of T-PLL.
Methods
Cells
A series of 47 T-PLL cases (TP followed by the case number) with frozen leukemic cells available was assembled from French Hematologic centers. All patients had major lymphocytosis. One T-PLL case (TP56) arose in a patient with ataxia telangiectasia. Diagnosis of T-PLL was established according to World Health Organization (WHO) classification of hematopoietic and lymphoid tumors.20 DNA samples were prepared from thawed tumor cells isolated on Histopaque gradient (Sigma-Aldrich, Saint Quentin Fallavier, France), and assessed for viability by trypan blue staining. T-PLL genomic DNAs were extracted using NucleoSpin Tissue kit (Macherey-Nagel, Hoerdt, France).
Allelotyping
Microsatellites and single nucleotide polymorphisms (SNPs) used are located in the 12p13 region. Primer sequences and percentage of heterozygosity were obtained from the GDB Human Genome Database,21 the UCSC Genome Bioinformatics,22 and the International HapMap Project.23 Microsatellite typing was achieved by polymerase chain reaction (PCR) using one fluorescent primer. SNP typing was done by PCR direct sequencing with ABI PRISM Big Dye Terminator v3.1 Ready Reaction Cycle sequencing kit (Applied Biosystems, Foster City, CA). Fluorescent products were analyzed using an ABI3130XL automated sequencer and the GeneMapper software v4.0 (Applied Biosystems). Primer sequences and PCR conditions are available upon request.
CDKN1B genomic sequencing
Coding regions and splicing junctions of CDKN1B were amplified from 50 ng genomic DNA in 50-μL reaction. The following primers were designed using Sequence Analysis 1.3.1 (Informagen, Greenland, NH) and Primer Express 1.0 (Applied Biosystems): CDKN1B-R1F: 5′-TTGATCAGCGGAGACTCG-3′; CDKN1B-R1R: 5′-CTCTTGCCACTCGTA-CTTGC-3′; CDKN1B-R2F: 5′-GAAGCACTGCAGAGACATGG-3′; CDKN1B-R2R: 5′-GCCAGGTAGCACTGAACACC-3′; CDKN1B-R3F: 5′-TATGGGGCCAACTTCTGC-3′; CDKN1B-R3R: 5′-TCCAGTGCGTGCTCCTTTAG-3′. PCR products were verified for size and amount on agarose gel, purified with NucleoFast 96 PCR plates (Macherey-Nagel), and sequenced. Analysis was performed using the Seqscape software (Applied Biosystems).
Quantitative real-time PCR
Total RNA was extracted using Trizol Reagent (Invitrogen, Frederick, MD). First-strand DNA synthesis was performed with 1 μg total RNA using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). For human studies, expression of CDKN1B was measured by quantitative real-time PCR (QRT-PCR) using 50 ng cDNA, and normalized to beta-glucuronidase (GUSB) expression, using TaqMan reagents (Hs00153277_m1 and 4326320E, respectively; TaqMan 2X Universal PCR Master Mix; Applied Biosystems). Results were analyzed with the Applied Biosystems 7500 System and v.1.3.1. associated software. Relative expression was expressed as 2−ΔΔCt in which ΔCt = CtCDKN1B − CtGUSB; ΔΔCt = ΔCtsample − ΔCtcalibrator.
Mice
Mice transgenic for the human MTPC1 open reading frame under the control of the CD2 regulatory sequences (MTCP1TG) were previously described.5 Cdkn1b+/− mice were generated by Koff and Coll (Kiyokawa et al24 ) and kindly obtained from W. Vainchenker's laboratory. Generation of the double mutant cohort and housing were done in the pathogen-free facility at the Institut Curie (Paris, France). Genotyping was done by PCR after weaning and verified at the end of the study. The following primers were used for Cdkn1b genotyping: kip1.5: 5′-AGCCCGAGCCTGGAGCGGATGGACGCC-3′, kipNeoRev: 5′-GGACATAGCGTTGGCTACCCGTGATATTGCTGA-3′, and kip1.3.2: 5′-CTCTCCACCTCCTGCCATTCGTATCTGCCC-3′. The following primers were used for genotyping the MTCP1 transgenics: CD2p13 F: 5′-TCTTGCTCTCTGTGTATGTGT-3′ and CD2p13 R: 5′-AAACTCGTCTCCTCTTCCAC-3′. Animals were followed as they aged and periodically controlled for hematologic status. At 14 months, animals were killed and analyzed. Two mice from the MTCP1WT/Cdkn1b+/− cohort developed splenomegaly due to erythroid metaplasia and were excluded from the study. For transplantation experiments, 2 million thawed splenocytes from animals with clonal T-cell expansions were resuspended in PBS, and intravenously injected in unirradiated histocompatible C57BL/6 mice. All experimental procedures were performed in strict accordance with the recommendations of the European Community (86/609/EEC) and the French National Committee (87/848) for care and use of laboratory animals. All animal experiments were carried out under the supervision of an investigator authorized by French Competent Veterinary Authority.
Histopathology
Organs were fixed in AFA (80% ethanol, 15% formaldehyde, 5% acetic acid) for 2 to 4 hours and further processed for paraffin embedding. Sections (4 μm each) were stained with hematoxylin eosin (HES) and analyzed by 2 different pathologists.
Flow cytometry analysis and sorting
Immunophenotyping of the mice was performed by flow cytometric analysis on single-cell suspensions prepared from blood or spleen, and stained with fluorescein isothiocyanate (FITC)–conjugated CD44; phycoerythrin (PE)–conjugated CD4; allophycocyanin (APC)–conjugated CD8; and biotin-conjugated CD122, Vβ2, Vβ5, Vβ6, Vβ8, Vβ9, Vβ10, Vβ11, Vβ13, and Vβ14 mouse antibodies. Bound biotinylated antibody was detected with PE-Cy5–conjugated streptavidin (all reagents from BD Pharmingen, Le Pont de Claix, France). Acquisitions were performed using a FACSDiva flow cytometer (BD Pharmingen), and results were analyzed with the FlowJo v.8.4.6 software (Tree Star, Ashland, OR). Sorting of clonal T-cell subpopulations from mice splenocytes according to their Vβ expression was performed on a FACSAria cell sorter (BD Pharmingen). CD8+ splenocytes were purified by negative depletion according to the manufacturer's recommendations (CD8a+ T Cell Isolation Kit; Miltenyi Biotec, Paris, France).
CDR3 spectratyping
CDR3 spectratyping used to define the clonality of the T-cell population was performed as described by Probert et al,25 but omitting the amplification step by nested PCR. Briefly, PCRs were performed using one of 2 different labeled Cβ primers (-HEX and -FAM) and one of the 20 unlabeled primers corresponding to the 20 Vβ segments, allowing amplification of all the murine Tcrb rearranged transcripts. Fluorescent fragments were analyzed as for allelotyping. A normal (Gaussian) distribution of the CDR3 length indicated a polyclonal repertoire. The presence of a highest peak representing more than 70% of all the peaks of the Gaussian curve was considered as indicative of a clonal population if shifted from the center of the curve obtained from the control.
Whole-cell extract preparation and immunoblotting analysis
Cells were collected by centrifugation at 600g for 5 minutes at 4°C and washed in cold PBS. Cell pellets were lysed by adding radioimmunoprecipitation assay (RIPA) buffer (100 mM Tris-HCl, 0.1 M NaCl, 1 mM EDTA, 1% Triton, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS]) supplemented with a cocktail of protease and phosphatase inhibitors, incubated on ice for 10 minutes, and centrifuged at 16 000g for 15 minutes at 4°C. Protein supernatants were collected, and equal amounts of protein (4 μg) were separated by SDS–polyacrylamide gel electrophoresis (PAGE, 12%) and then electrotransferred to nitrocellulose membrane. Nonspecific antibody-binding sites were blocked by incubation in blocking buffer (PBS, 0.1% Tween 20 [PBS-T] with 5% wt/vol nonfat dry milk) for 1 hour at room temperature. After PBS-T washes, membranes were incubated overnight at 4°C with p27Kip1 (BD Pharmingen) and β-actin (ac-74; Sigma-Aldrich) antibodies diluted according to the manufacturer's instructions in PBS-T with 5% BSA. After washes with PBS-T, membranes were incubated with horseradish peroxidase–conjugated secondary antibody diluted in blocking buffer for 1 hour at room temperature. Proteins were detected by incubating the membrane with Super Signal West Pico (Pierce, Rockford, IL) detection reagent. p27Kip1 expression level was analyzed, quantified, and normalized to β-actin expression, using a luminescent image analyzer (LAS-1000plus; Fujifilm, Saint Quentin en Yvelines, France).
Results
12p13 deletion in human T-PLL
An earlier attempt to determine genes or genomic regions altered in T-PLL disease using allelotyping identified a 12p13 deletion in 43% of T-PLL cases, including a case with a biallelic deletion. The minimal region of deletion (MRD) spanned approximately 1 megabase (Mb), between the markers b312C2T7 and D12S320.11
To identify the gene targeted by the 12p13 deletion, we extended our initial study by microsatellite and single nucleotide polymorphism (SNP) allelotyping on a series of 23 T-PLL cases (including the 21 cases from the initial study) (Table 1). Eleven (48%) of 23 patients showed loss of heterozygosity (LOH) of at least one marker in the 12p13 region, and data obtained from 2 cases further reduced the MRD from approximately 1 Mb to 216 kilobases (kb). Results obtained from case TP42 delineated the centromeric boundary of the deletion with D12S1581. Two polymorphisms located within the CDKN1B gene were found to be the only markers with a LOH pattern in case TP21, characterizing the telomeric boundary of the deletion at SNP rs3759216. This 216-kb region (chr12: 12 759 353-12 975 414, NCBI Build 36.1) included 6 genes or coding sequences (CDKN1B, APOLD1, DDX47, RAIG1, GPRC5A, and BC062343) among which CDKN1B represented a plausible candidate gene, because it encodes the cyclin-dependent kinase inhibitory protein p27KIP1.
LOH analysis of 23 paired germline and leukemic human DNA samples
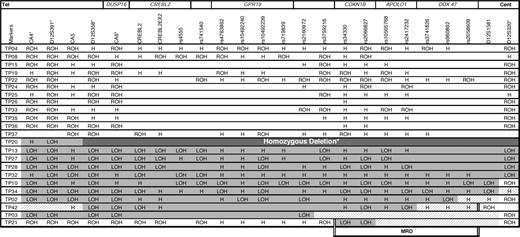
ROH indicates retention of heterozygosity (ie, heterozygosity with a balanced representation of the 2 alleles); LOH, loss of heterozygosity; H, homozygous; Tel, telomere; Cent, centromere; and MRD, minimal region of deletion. Regional status is indicated as follows: white square, ROH; light grey square, LOH; dark grey square, homozygous deletion; hatched square, undefined status. On the top is the approximate location of genes in relation to the markers. Double vertical lines correspond to the boundaries of the minimal region of deletion. *Results previously reported.11
We determined the CDKN1B genomic sequence in a series of 47 T-PLL cases. DUSP16, which encodes a dual specificity phosphatase,26 and CREBL2, located in the vicinity of the MRD, were also sequenced in some cases. No mutation was found in these 2 latter genes. Interestingly, a nonsense mutation (c.118G>T/p.E40X described by coding DNA reference sequence BC001g71) in CDKN1B was detected in one of 47 cases analyzed by sequencing (Figure 1A). This case (TP72) had no evidence of 12p13 deletion.

Genomic and expression analyses of CDKN1B in human T-PLL samples. (A) Sequence analysis of CDKN1B exon 1 amplified from the TP72 DNA leukemic sample. This patient carried a c.118G>T/p.E40X nonsense mutation in one allele. Chromatogram is shown on the bottom panel; sequence is indicated above. K=G+T. Putative translation of both alleles is shown on the upper panel using the single letter code. X indicates the stop codon. (B) Expression analysis of 20 T-PLL cases and of CD3+ T cells from 5 healthy individuals. Means are indicated as empty squares; plus or minus SDs are indicated as lines. An asterisk indicates the significance at P < 10−3 of the between-group analysis (bracket) determined by Student t test. CD3+ indicates control CD3+ T cells; 12pDEL, patients with 12p13 deletion; and 12pWT, patients without evidence of 12p13 genomic alteration. CDKN1B relative expression is expressed as 2− ΔΔCt in which ΔCt = CtCDKN1B − CtGUSB; ΔΔCt = ΔCtsample − ΔCtcalibrator.
Genomic and expression analyses of CDKN1B in human T-PLL samples. (A) Sequence analysis of CDKN1B exon 1 amplified from the TP72 DNA leukemic sample. This patient carried a c.118G>T/p.E40X nonsense mutation in one allele. Chromatogram is shown on the bottom panel; sequence is indicated above. K=G+T. Putative translation of both alleles is shown on the upper panel using the single letter code. X indicates the stop codon. (B) Expression analysis of 20 T-PLL cases and of CD3+ T cells from 5 healthy individuals. Means are indicated as empty squares; plus or minus SDs are indicated as lines. An asterisk indicates the significance at P < 10−3 of the between-group analysis (bracket) determined by Student t test. CD3+ indicates control CD3+ T cells; 12pDEL, patients with 12p13 deletion; and 12pWT, patients without evidence of 12p13 genomic alteration. CDKN1B relative expression is expressed as 2− ΔΔCt in which ΔCt = CtCDKN1B − CtGUSB; ΔΔCt = ΔCtsample − ΔCtcalibrator.
To evaluate the consequences of 12p13 deletion onto CDKN1B expression, quantitative RT-PCR (QRT-PCR) was performed on a series of 20 T-PLL cases with known genomic status and available RNA (Figure 1B). CDKN1B expression of the 10 cases with 12p13 deletion was approximately half of the expression of normal CD3+ peripheral lymphocytes (relative expression; 2−ΔΔCt = 0.47 ± 0.13 vs 1.04 ± 0.19, respectively; P < .001). CDKN1B expression was more heterogeneous in cases without 12p13 deletion, with 6 of 10 cases with expression similar to that of normal CD3+, and the 4 other cases with a reduced expression similar to that of T-PLL with 12p13 deletion (Figure 1B).
Taken as a whole, these data define a small minimal region of 12p13 deletion in which CDKN1B is located. This region was deleted in 11 (48%) of 23 cases studied. In addition, a nonsense mutation of CDKN1B in one case strongly suggested that this gene was the target of the 12p13 deletion in T-PLL. Furthermore, 4 (40%) of 10 cases without detected genomic alteration had a reduced CDKN1B expression of unknown origin. However, persistence of CDKN1B expression in most T-PLL cases excluded CDKN1B as a classical tumor suppressor gene, and suggested that CDKN1B haploinsufficiency may be the oncogenetic event driven by the 12p13 deletion. We thus tested this hypothesis using animal models.
Early expansion of CD8 memory lymphocytes in Cdkn1b+/−/MTCP1TG mice
We generated a cohort of mice heterozygous for Cdkn1b inactivation and transgenic for MTCP1 (DM for double mutant) and compared it with cohorts with the other genotypes obtained from the genetic crosses, that is, Cdkn1b+/+/MTCP1TG (Tg for transgenic), Cdkn1b+/−/MTCP1WT (Htz for Cdkn1b heterozygote), and Cdkn1b+/+/MTCP1WT (WT for wild type) mice. CD8 memory T-cell (CD8m) population was evaluated during aging, since expansion of this population was characterized in MTCP1 transgenics before the emergence of leukemia.7 Indeed, CD8m accumulated more rapidly in MTCP1TG than in nontransgenic animals. However, this phenomenon was more dramatic in DM mice (Figure 2A,B). In blood samples obtained at 7 months, the percentage of CD8m among CD8 T cells was 42.7% (± 19.1%), 29.2% (± 10.5%), 25.1% (± 9.7%), and 18.4% (± 4.7%) for the DM, Tg, Htz, and WT mice, respectively (Figure 2B). DM mice had higher levels of CD8m compared with mice with the other genotypes, but this difference was significant only in comparison to nontransgenic animals (vs Htz: P = .023; vs WT: P = .003). At 14 months, the accumulation of CD8m in the DM mice (87.6% ± 9.2%) was significantly higher than in mice with the 3 other genotypes (vs 62.1% ± 7.6%, P < .001; vs 45.3% ± 11.9%, P < .001; and vs 26.1% ± 4.0%, P < .001; for Tg, Htz, and WT mice, respectively) (Figure 2B).

FACS analyses of blood samples during aging. (A) Memory CD8 T-cell accumulation in representative animals from the 4 genotypes at 10 months. DM indicates Cdkn1b+/−/MTCP1TG; Tg, MTCP1 transgenics; Htz, Cdkn1+/−; and WT, wild type. (Left) Dot plots of CD4-PE and CD8-APC gated on the lymphocyte population and (right) CD44-FITC and CD122-PE-Cy5 gated on the CD8+ cells. (B) Percentage of CD8m (CD8+CD44HICD122HI) among total CD8 (CD8T) cells at 7, 10, and 14 months in mice with the 4 different genotypes (DM represented by black diamond; Tg, black circle; Htz, empty diamond; and WT, empty circle). Means are indicated as empty squares; ± SDs are indicated as lines. Significance of between-group analyses (brackets) determined by Student t test is indicated as follows: NS, nonsignificant difference; *P < 5.10−2; **P < 5.10−3; and ***P < .001. At 10 months, a group of 4 DM mice with higher CD8m accumulation is identified by a rectangle. (C) Vβ repertoire in CD8+ cells in the different genotypes at 10 months. The distribution of 9 TCR Vβ in CD8m (CD8+CD44HI) is represented by black rectangle; and in CD8n (CD8+CD44LOW) by gray rectangle, in representative animals from 1 wild-type (WT1), 1 MTCP1 transgenic (TG10), and 4 DM mice.
FACS analyses of blood samples during aging. (A) Memory CD8 T-cell accumulation in representative animals from the 4 genotypes at 10 months. DM indicates Cdkn1b+/−/MTCP1TG; Tg, MTCP1 transgenics; Htz, Cdkn1+/−; and WT, wild type. (Left) Dot plots of CD4-PE and CD8-APC gated on the lymphocyte population and (right) CD44-FITC and CD122-PE-Cy5 gated on the CD8+ cells. (B) Percentage of CD8m (CD8+CD44HICD122HI) among total CD8 (CD8T) cells at 7, 10, and 14 months in mice with the 4 different genotypes (DM represented by black diamond; Tg, black circle; Htz, empty diamond; and WT, empty circle). Means are indicated as empty squares; ± SDs are indicated as lines. Significance of between-group analyses (brackets) determined by Student t test is indicated as follows: NS, nonsignificant difference; *P < 5.10−2; **P < 5.10−3; and ***P < .001. At 10 months, a group of 4 DM mice with higher CD8m accumulation is identified by a rectangle. (C) Vβ repertoire in CD8+ cells in the different genotypes at 10 months. The distribution of 9 TCR Vβ in CD8m (CD8+CD44HI) is represented by black rectangle; and in CD8n (CD8+CD44LOW) by gray rectangle, in representative animals from 1 wild-type (WT1), 1 MTCP1 transgenic (TG10), and 4 DM mice.
Although generally higher than in mice with the other genotypes, accumulation of CD8m in DM mice was noticeably variable among individual animals. In particular, at 10 months, 2 groups could be distinguished: one superimposable with that of Tg mice and one with a more dramatic accumulation of CD8m T cells (Figure 2B). A possible explanation was the emergence of clonal populations contributing to the CD8m expansion in the second group of DM mice. To test such a hypothesis, the Tcr Vβ repertoire of this population was studied by flow cytometry using a set of 9 anti-Vβ antibodies recognizing approximately 80% of the Tcr repertoire (Figure 2C). An unskewed repertoire was defined when the Vβ distribution in CD8m cells was superimposable with that of the naive CD8 (CD8n) population. Such a repertoire was observed in Htz, Tg, and WT mice, including the Tg mouse with more than 40% of CD8m cells, confirming our previous report.7 Contrasting with these observations, skewed repertoires were found in half of the DM mice analyzed (4/8), providing indirect (dramatic underrepresentation of the 9 Vβ tested in DM3 and DM4) or direct (overrepresentation of Vβ2 and Vβ5 in DM8, and of Vβ8 in DM1) evidence of clonal or oligoclonal expansions. Interestingly, these skewed repertoires were revealed in the 4 DM mice with the highest percentages of CD8m cells.
Nature of the cellular expansions in Cdkn1b+/−/MTCP1TG mice
Despite early emergence of clonal expansions in DM mice, no spontaneous death or signs of malignancy were detected in the cohort at 14 months, the age at which animals were killed. Macroscopic examination of the mice was unremarkable, with the exception of mild splenomegaly (more than 150 mg) in 5 DM animals, and macronodular and enlarged liver in 2 DM animals (Figure 3A).

Analysis of the animals at 14 months of age. (A) Characteristics of the 4 genotypes at the age of killing. DM indicates Cdkn1b+/−/MTCP1TG; Tg, MTCP1 transgenics; Htz, Cdkn1+/−; WT, wild type; WBC, white blood cell count; CD8m/CD8T, ratio of CD8 memory cells (CD8+CD44HI) to total CD8+ splenocytes determined by FACS; CD8m/T-cells, ratio of CD8 memory cells (CD8+CD44HI) to total T-cell splenocytes (CD4+ and CD8+ lymphocytes) determined by FACS; CD4/CD8, ratio of CD8+ splenocytes to CD4+ splenocytes determined by FACS; §, 2 mice developed an erythroid metaplasia and were excluded from the study; *, significantly higher by Student t test in DM mice compared with the 3 other cohorts with P < .05; and **, significantly lower by Student t test in DM mice compared with the 3 other cohorts with P < .05; in parentheses is indicated the number of mice analyzed for WBC. (B) Histopathological analysis after HES staining of liver and spleen from characteristic mice for each genotype. Images were acquired with a DMRB microscope with 10×/0.30 and 20×/0.50 PL Fluotar objective lenses (Leica, Rueil-Malmaison, France) equipped with a KY-F50 Tri CCD camera (JVC, Clara Vision, Massy, France) and analyzed with Vega v2.0 software (Clara Vision) without further image processing. The lymphocytic nodules are indicated by arrows. Magnifications are indicated.
Analysis of the animals at 14 months of age. (A) Characteristics of the 4 genotypes at the age of killing. DM indicates Cdkn1b+/−/MTCP1TG; Tg, MTCP1 transgenics; Htz, Cdkn1+/−; WT, wild type; WBC, white blood cell count; CD8m/CD8T, ratio of CD8 memory cells (CD8+CD44HI) to total CD8+ splenocytes determined by FACS; CD8m/T-cells, ratio of CD8 memory cells (CD8+CD44HI) to total T-cell splenocytes (CD4+ and CD8+ lymphocytes) determined by FACS; CD4/CD8, ratio of CD8+ splenocytes to CD4+ splenocytes determined by FACS; §, 2 mice developed an erythroid metaplasia and were excluded from the study; *, significantly higher by Student t test in DM mice compared with the 3 other cohorts with P < .05; and **, significantly lower by Student t test in DM mice compared with the 3 other cohorts with P < .05; in parentheses is indicated the number of mice analyzed for WBC. (B) Histopathological analysis after HES staining of liver and spleen from characteristic mice for each genotype. Images were acquired with a DMRB microscope with 10×/0.30 and 20×/0.50 PL Fluotar objective lenses (Leica, Rueil-Malmaison, France) equipped with a KY-F50 Tri CCD camera (JVC, Clara Vision, Massy, France) and analyzed with Vega v2.0 software (Clara Vision) without further image processing. The lymphocytic nodules are indicated by arrows. Magnifications are indicated.
Immunologic analysis of the splenocytes was in agreement with data obtained from blood samples during aging. The proportion of CD8m T cells among the total number of CD8 T cells (CD8T) was significantly higher in DM mice (87.6% ± 9.2%) compared with control cohorts (vs 62.1% ± 7.6%, P < .001; vs 45.3% ± 11.9%, P < .001; and vs 26.1% ± 4.0%, P < .001; in Tg, Htz, and WT mice, respectively). The proportion of CD8m T cells among T lymphocytes was also significantly higher in DM mice (59.5% ± 17.2%) compared with control cohorts (vs 30.2% ± 14.5%, P < .001; vs 16.8% ± 7.1%, P < .001; and vs 10.0% ± 4.6%, P < .001; in Tg, Htz, and WT mice, respectively). This increase in memory CD8 T cells was dramatic and at the expense of the other cell populations, as shown by the inversion of the ratio CD4/CD8 in DM mice (Figure 3A). Histopathological examination revealed lymphoid organs with a disorganized architecture in most DM mice (Figure 3B). However, the most striking feature was the consistent presence of lymphocytic nodules in the liver of these animals, either with a multiple micronodular pattern (5/10), or as few large lymphocytic nodules (5/10), visible at the macroscopic examination. This lymphoid infiltration was generally concentrated around the central-lobular veins. Other organs such as the kidneys were occasionally infiltrated with lymphoid cells. In comparison, no or minimal lymphoid infiltrates were observed in the liver of animals with other genotypes, including Tg mice and the Htz mouse with a high CD8m percentage (Htz10).
Because Vβ fluorescence-activated cell sorting (FACS) patterns were compatible with monoclonal or oligoclonal populations in DM mice, clonality was evaluated by a sensitive technique, the CDR3 spectratyping.25,27 In this technique, T-cell diversity is evaluated according to the CDR3 size pattern generated by the Tcrb V(D)J recombination, analyzing rearrangements using each of the 20 Vβ segments separately. Patterns were examined for each Vβ, and the presence of a clone was determined according to the presence of a highest peak representing more than 70% of all the peaks of the Gaussian curve. Moreover, a shift of the major peak from the center of the curve was indicative of a clonal event (Figure 4A). For rarely used Vβ segments, the detection of a single peak when no significant signal was detected in WT mice indicated the presence of a clonal population. According to these criteria, no clonal T-cell population was detected at 14 months in the WT and Tg mice (2 individual mice evaluated for each cohort). Conversely, all the DM mice tested (6 animals total) revealed multiple emergences of T-cell clones (range: 2 to 7; mean: 3.5 clones per animal) (Figure 4B). In 3 cases, clonality was also confirmed by Southern blotting analysis (data not shown). Interestingly, in one Htz animal (Htz10) of the 4 tested, CDR3 spectratyping revealed a single Vβ5 clonal population. This mouse was characterized by a high CD8m/CD8T ratio at 14 months, but a lower expansion of these cells compared with DM mice, without inversion of the CD4/CD8 ratio.

Multiple emergence of leukemic clones in Cdkn1b+/−/MTCP1TG mice. (A) The presence of clonal populations was detected by CDR3 spectratyping. Examples of chromatograms are shown for different mice from the 4 genotypes, and different Vβ segments used for Tcrb rearrangements. Black rectangles indicate signal saturation. Arrows indicate rearrangements fulfilling criteria for clonality. DM indicates Cdkn1b+/−/MTCP1TG; Tg, MTCP1 transgenics; Htz, Cdkn1+/−; and WT, wild type. (B) Summary of spectratyping data from the 14 mice tested. P indicates polyclonal pattern; C, clonal pattern; U, unknown, no significant signal; and NA, not available.
Multiple emergence of leukemic clones in Cdkn1b+/−/MTCP1TG mice. (A) The presence of clonal populations was detected by CDR3 spectratyping. Examples of chromatograms are shown for different mice from the 4 genotypes, and different Vβ segments used for Tcrb rearrangements. Black rectangles indicate signal saturation. Arrows indicate rearrangements fulfilling criteria for clonality. DM indicates Cdkn1b+/−/MTCP1TG; Tg, MTCP1 transgenics; Htz, Cdkn1+/−; and WT, wild type. (B) Summary of spectratyping data from the 14 mice tested. P indicates polyclonal pattern; C, clonal pattern; U, unknown, no significant signal; and NA, not available.
To assess the malignant nature of clonal expansion of the Cdkn1b+/−/MTCP1TG mice, engraftment of lymphocytes in histocompatible immunocompetent animals was attempted with 3 DM splenocytes. In 2 of 3 cases, CD8m expansion was evidenced after 3 months of engraftment, and CDR3 spectratyping revealed patterns similar to that of the animals of origin. In these 2 cases, direct Tcrb cDNA sequencing identified identical clonal V(D)J rearrangements in the DM mouse and in the engrafted host, demonstrating the capability of these expansions to graft into immunocompetent hosts (data not shown). However, as in 14-month-old DM mice, engrafted animals had no clinical symptom of leukemic disease.
Altogether, CD8m expansions found in DM mice have some criteria of leukemic disease, such as clonal nature, enlarged cells in forward/scatter FACS analyses (data not shown), invasion of lymphoid and nonlymphoid tissues, and capability to graft into murine host. However, in absence of clinical illness, despite a long observation period, these expansions would be better defined as preleukemic populations than as frank leukemia.
Cdkn1b haploinsufficiency in clonal CD8 cells from Cdkn1b+/−/MTCP1TG mice
To evaluate if haploinsufficiency suggested from molecular studies in human T-PLL was also verified in the animal model, the status of the second allele of Cdkn1b was investigated in preleukemic clonal populations from DM mice. Clonal CD8m splenocytes from 2 DM mice (DM1 and DM8) were sorted out based on their Vβ expression, characterized for Cdkn1b gene copy number and sequencing. No deletion or mutation of the Cdkn1b wild-type allele was detected (data not shown). Furthermore, p27Kip1 expression was quantified by immunoblotting in these sorted clonal populations and compared with that of CD8+ lymphocytes from the different mouse genotypes. Indeed, Cdkn1b+/− CD8+ cells expressed approximately half the p27Kip1 as did Cdkn1b+/+ CD8+ cells (Figure 5). Confirming the absence of evidence for somatic alteration of the wild-type allele, p27Kip1 expression in preleukemic clonal populations was similar to that of polyclonal CD8+ T-cell populations from DM mice (Figure 5). These results demonstrate that Cdkn1b haploinsufficiency accelerates leukemogenesis in a mouse model of T-PLL.

Clonal preleukemic cells expressed p27Kip1. p27Kip1 expression was determined by Western blotting and normalized to β-actin expression. (A) Western blot of purified polyclonal CD8+ T cells of 2 mice of each genotype, and of 2 clonal preleukemic CD8m T cells from 2 DM mice at 14 months, sorted based on their Vβ expression (DM1 and DM8). DM indicates Cdkn1b+/−/MTCP1TG; Tg, MTCP1 transgenics; Htz, Cdkn1+/−; and WT, wild type. (B) Relative p27Kip1 expression normalized to β-actin expression, and to mean expression of wild-type animals. Three mice of each genotype and the 2 sorted clonal populations were analyzed. Results obtained from polyclonal populations were pooled according to the Cdkn1b status. Significance of between-group analyses (brackets) determined by Student t test is indicated as follows: NS, nonsignificant difference; *P = .02; **P = .002.
Clonal preleukemic cells expressed p27Kip1. p27Kip1 expression was determined by Western blotting and normalized to β-actin expression. (A) Western blot of purified polyclonal CD8+ T cells of 2 mice of each genotype, and of 2 clonal preleukemic CD8m T cells from 2 DM mice at 14 months, sorted based on their Vβ expression (DM1 and DM8). DM indicates Cdkn1b+/−/MTCP1TG; Tg, MTCP1 transgenics; Htz, Cdkn1+/−; and WT, wild type. (B) Relative p27Kip1 expression normalized to β-actin expression, and to mean expression of wild-type animals. Three mice of each genotype and the 2 sorted clonal populations were analyzed. Results obtained from polyclonal populations were pooled according to the Cdkn1b status. Significance of between-group analyses (brackets) determined by Student t test is indicated as follows: NS, nonsignificant difference; *P = .02; **P = .002.
Discussion
Deletion in the 12p13 region was previously shown to be a frequent event in T-PLL.11 Here we provided a series of results supporting that CDKN1B is the target gene of this deletion. First we completed a LOH study using microsatellite and single nucleotide polymorphism (SNP) analysis for a denser coverage of the region, and herein refined the previous minimal region of deletion (MRD) to a segment of only 216 kb. This region was included in the MRD previously defined in other types of leukemias,28,–30 but was excluded from the MRD defined by Baens et al.31 These data strongly suggest that heterogeneity in 12p13 deletions exists among different types of hematologic disorders, implicating different putative target genes such as ETV6 or DUSP16.26,32
Six genes or coding sequences were located in the MRD defined here, among which was CDKN1B, previously shown to play a clear role in oncogenesis. Deletion or underexpression of CDKN1B is a common feature of human malignancies, including both solid tumors and hemopathies (for review, see Slingerland and Pagano18 ). Deleterious somatic mutations in CDKN1B have been rarely reported.33,–35 To our knowledge, the only nonsense somatic mutation ever reported for CDKN1B was located at codon 76 in an adult T-cell leukemia/lymphoma.33 The same mutation was recently reported in the germ line of a family with multiple endocrine neoplasia syndrome (MEN1).36 Interestingly, by sequencing CDKN1B in a large series of T-PLL, we identified another nonsense mutation at codon 40 in one case carrying no evidence of 12p13 deletion. The identification of this mutation further supported that CDKN1B could be the target gene for the 12p13 deletion in T-PLL.
Genomic alterations of CDKN1B were found in approximately half of the T-PLL cases studied here. Furthermore, the CDKN1B expression study showed that a significant number of T-PLL cases (4 of 10 studied) without evidence of 12p13 deletion have a decreased level of CDKN1B transcripts, raising the possibility that other mechanisms of inactivation also occur in T-PLL.
All T-PLL cases with 12p13 deletions or mutation had monoallelic CDKN1B alterations, except one case with a biallelic deletion. As expected, expression of this gene in leukemic cells was found to be approximately half of that found in normal counterpart lymphocytes, excluding possible undetected biallelic deletions or transcriptional repression of the second allele. CDKN1B gene belongs to an unusual class of tumor suppressor genes that do not conform to the Knudson “2-hit” model.37 However, CDKN1B copy number gene reduction or underexpression has previously been demonstrated to play a role in oncogenesis.12 This hypothesis, which arose from human tumor analyses, was confirmed using elegant animal models. Fero et al showed that Cdkn1b heterozygous mice are tumor prone in response to γ-irradiation or chemical carcinogenesis, without affecting the wild-type Cdkn1b allele in tumors. The phenotype of Cdkn1b+/− mice was intermediate between wild-type mice and Cdkn1b−/− mice. Thus in this mouse model, p27Kip1 acted as a tumor suppressor in a dose-dependent manner.19 In contrast, in cellular models of mammary carcinogenesis expressing ErbB2/Neu or cyclin D1, increased proliferation was shown in a Cdkn1b+/− background but impaired in Cdkn1b−/− cells.38 The specific consequence of CDKN1B alterations with regard to tumor progression may be attributed to the complicated role of its product on the cell cycle, depending on its cellular concentration.13
We generated a mouse model of human T-PLL by crossing MTCP1TG mice with Cdkn1b+/− mice to search for a putative haploinsufficient role of Cdkn1b in this disease. The consistent emergence of T-cell clonal preleukemic expansions in double mutant mice strongly supported Cdkn1b as an important player in leukemogenesis. It should be noticed that in their present pathogen-free housing environment, MTCP1 transgenics had no clinical leukemic disease even after 2 years of life. Similar to results from analyses of the human T-PLL, the persistence of a functional wild-type allele in murine leukemic populations indicated that copy number reduction of CDKN1B was sufficient for leukemogenesis. Identification of only one patient with a biallelic deletion precluded a conclusion of whether alteration of the second allele is neutral or further contributes to the malignant process. Of note, preliminary data obtained from a few MTCP1 transgenic animals in a Cdkn1b−/− background did not show a faster CD8m accumulation than in a Cdkn1b+/− background (E.L.T. and M.-H.S., unpublished data, June 2007).
We took advantage of the possibility to investigate clonality in T-cell population to demonstrate early and multiple emergences of clonal preleukemic CD8m populations in double mutant mice. The mechanism by which Cdkn1b haploinsufficiency favors initiation of clonal events remains unclear and will merit further investigations. The fact that human T-PLL cells have lower p27KIP1 expression favors a cell-autonomous mechanism. However, a role of the stromal cells (cell nonautonomous) in Cdkn1b+/− animals was clearly demonstrated for hematopoietic homeostasis,39 and cannot be excluded in our model.
Interestingly, one Cdkn1b+/− nontransgenic animal developed a clonal preleukemic expansion, raising the possibility that this event could predispose to T-PLL. Rare patients with a variant of MEN1 syndrome were reported to bear a monoallelic mutation of CDKN1B.36 Although not yet reported, our data may suggest an increased incidence of leukemia in this variant syndrome.
Altogether our results strongly support an important role for CDKN1B haploinsufficiency in T-PLL. Alteration of a cell-cycle inhibitor is not unexpected in such an aggressive form of leukemia. Interestingly, consistent inactivation of another cell-cycle inhibitor, CDKN2A, is found in T-cell acute lymphoblastic leukemia.40 However, no obvious clinical difference was demonstrated between patients with or without genetic alteration of CDKN1B (E.L.T. and M.-H.S., unpublished data, June 2007), raising the possibility that other mechanisms impairing cell cycle regulation remain to be identified in T-PLL cases without a 12p13 deletion.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
We thank Evelyne Lauret and William Vainchenker for kindly providing the Cdkn1b knock-out animals, Andrew Koff for generously authorizing their use, Isabelle Grandjean and her team for expertise with animals, Anne-Flore Albertini and Sylvia Carvalo for assistance, and Dan Williamson, Jean Soulier, and Martine F. Roussel for critical reading of the paper. We are indebted to the French hematologists who provided patient samples: B. Cazin (Lille); R. Garand (Nantes); M. J. Grange, V. Leblond, F. Nguyen Khac, H. Merle-Beral, F. Valensi, B. Varet, I. Radford-Weiss, O. Hermine, R. Delarue, V. Levy, J. C. Brouet, P. Rousselot (Paris); G. Damaj (Creteil); X. Troussard (Caen); S. Daliphard, P. Cornillet (Reims); D. Lusina (Aulnay); K. Ghomari (Beauvais); C. Bertout (Brest); O. Tournilhac (Clermond-Ferrand); M. Maynadie (Dijon); V. Izydirczyk (Le Havre); E. Callet-Bauchu, B. Coffier (Lyon); and F. Lellouche (Quimper).
This work was supported by grants from the Institut National du Cancer (INCa), Inserm and Institut Curie, Section de Recherche. This work is part of the “Canceropole Ile-de-France–Mouse models of human cancer” program coordinated by M. Giovannini. E.L.T. and M.J. are recipients of Fellowships from the Ministère de la Recherche et de la Technologie (MRT). G.D. is a recipient of grants from the Canceropole Ile-de-France, the Fondation de France, and the Association Pour la Recherche sur l'Ataxie-Télangiectasie (APRAT).
Authorship
Contribution: E.L.T. performed research, analyzed data, and wrote the paper; M.J., G.D., G.P., and N.G. performed research and analyzed data; D.B. performed research; A.V.-S. analyzed pathological specimens; and M.-H.S. designed research and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Marc-Henri Stern, Inserm U830, Institut Curie, Centre de Recherche, 26 rue d'Ulm, 75248 Paris cedex 05, France; e-mail: marc-henri.stern@curie.fr.
