

Abstract
Nucleophosmin (NPM) is a multifunctional phosphoprotein that acts as nucleocytoplasmic shuttling protein, with tumour suppressor and oncogenic functions. Recently, NPM mutations have been found in a subset of adults with de novo acute myeloid leukemia (AML). These mutations occur in the last coding exon (exon 12), causing a frameshift and the formation of novel C-termini. The abnormal mutated NPM protein shows aberrant cytoplasmic localization and is frequently associated with FLT3 mutation.
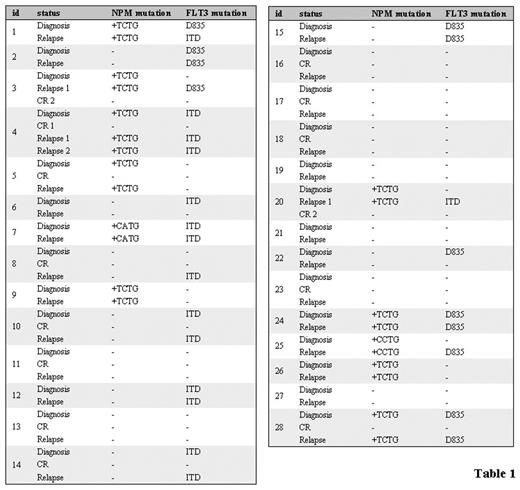
These observations provide basis for studies of the pathogenesis in AML. We did sequential analysis on patient samples during the clinical course to investigate the stability and pathogenetic role of NPM mutation in AML and the association with FLT3 mutations. The NPM mutations were determined by D-HPLC analysis; samples exhibiting an abnormal D-HPLC profile were confirmed by direct sequencing. We investigated 28 patients of de novo AML. Eleven samples were sequenced since they showed an heteroduplex D-HPLC profile. Type A mutation (960_963dupTCTG) was the commonest observed change, occurring in 9/11 samples, followed by type B mutation (960_963insCATG) in 1 case and type D mutation (960_963insCCTG) in 1 case. Furthermore, we observed that 5/11 patients harboring NPM mutation presented also mutant FLT3 at diagnosis. Analyzing NPM mutations during progression of disease, we observed that NPM mutation disappeared at complete remission and the same mutation reappeared at relapse. No differences were found in wild type NPM.
Instead, we found a modification of FLT3 status associated to evolution of disease in 7/28 patients: 2 patients lost the mutation at relapse, 4 patients acquired the mutation at relapse and 1 patient modified the mutation from D835 to ITD (Table 1). Together these results suggest that NPM mutations and not FLT3 mutations may have utility as a potential marker for monitoring minimal residual disease. Studies on the biological effects of NPM mutations will contribute to disclose the role of NPM mutations in the pathogenesis of AML and their interactions with other genetic alterations such as FLT3.
Disclosure: No relevant conflicts of interest to declare.
Acknowledgments: COFIN 2005 (Myelodisplastic syndromes: pathogenetic models and promise of new therapies),COFIN 2003 (Molecular therapy of leukemias), by FIRB 2001, by the University of Bologna (60%), by the Italian Association for cancer research (A.I.R.C.), by the Italian National Research Council (C.N.R), by Fondazione Del Monte of Bologna e Ravenna (Italy) and A.I.L. grants.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal