Abstract
Hereditary hemochromatosis (HH) is a common autosomal recessive genetic disorder of iron metabolism. The HFE candidate gene encoding an HLA class I-like protein involved in HH was identified in 1996. Two missense mutations have been described: C282Y, accounting for 80% to 90% of HH chromosomes, and H63D, which is associated with a milder form of the disease representing 40% to 70% of non-C282Y HH chromosomes. We report here on the analysis of C282Y, H63D, and the 193A→T substitution leading to the S65C missense substitution in a large series of probands and controls. The results confirm that the C282Y substitution was the main mutation involved in hemochromatosis, accounting for 85% of carrier chromosomes, whereas the H63D substitution represented 39% of the HH chromosomes that did not carry the C282Y mutation. In addition, our screening showed that the S65C substitution was significantly enriched in probands with at least one chromosome without an assigned mutation. This substitution accounted for 7.8% of HH chromosomes that were neither C282Y nor H63D. This enrichment of S65C among HH chromosomes suggests that the S65C substitution is associated with the mild form of hemochromatosis.
HEREDITARY hemochromatosis (HH) is an autosomal recessive disease affecting iron metabolism and is commonly found in whites. Populations of northern European origin show the highest frequency of HH, with 1 in 300 individuals affected.1,2 HH is characterized by increased iron absorption and progressive iron storage that results in organ damage. The clinical consequences of iron overload can be prevented by early diagnosis and treatment with repeated venesections. Because early diagnosis during the asymptomatic phase would be more effective for preventive treatment, a reliable genetic-based test is required. TheHFE gene was identified by positional cloning in 1996,3 and relevant studies have been performed that support HFE as being the gene responsible for hemochromatosis. The HFE gene encodes an HLA class I-like protein that, in association with β2-microglobulin, has an expression pattern correlated with the localization of iron absorption.3-5 HFE seems to play a role in iron uptake by interacting with the transferrin receptor (TfR), leading to a decreased affinity of TfR for transferrin.6-8
The single-base substitution 845G→A (C282Y) has been well characterized as being the main mutation responsible for hemochromatosis in all the population studies performed throughout the world. It accounts for 80% to 90% of HH chromosomes, with 81% to 90% of HH patients being homozygous for C282Y.3,9-14 The functional relevance of the C282Y mutation has been clarified by cell transfection assays showing that this mutation abolishes a conserved cysteine that impairs β2-microglobulin association and cell surface expression of the HFE protein.4,15 Hence, the C282Y mutation eliminates the interaction with TfR that may be sufficient to cause iron storage overload.7
Correlations between phenotype and genotype have also shown that nonhomozygous probands for C282Y have lower iron overload than C282Y homozygous subjects according to their iron status marker.10,12,13 It has been reported that, among non-C282Y HH chromosomes, 21% to 43%11-13,16 and possibly as many as 64% to 73%, according to the last estimation from pooled studies,14,17 bear the H63D (187C→G) substitution, indicating that the H63D frequency is increased among HH probands. Compound heterozygotes C282Y/H63D account for approximately 7% of hemochromatosis subjects, and the lower than expected frequency of H63D homozygotes found among HH probands compared with the H63D frequency in the population is evidence of the incomplete penetrance of the H63D mutation. Thus, the H63D substitution may be considered as a genetic variant that increases the risk of developing a mild form of hemochromatosis.11-14,16,17 The functional significance of H63D was recently shown in a cell transfection assay in which this substitution led to a loss of ability to reduce the TfR affinity for transferrin compared with wild-type HFE.7
Several studies, including the systematic sequencing of the coding region performed on a total of 44 HH chromosomes that do not bear C282Y or H63D in European and North American patients,11,13,18have been performed on hemochromatosis subjects from different populations. Despite these reports, no other obvious mutation for HH, except for a single delC mutation,19 have been described. Several polymorphisms have also been reported.18,20,21These are predominantly localized in intron sequences except for one, namely 193A→T, which leads to a serine-to-cysteine substitution (S65C) localized in exon 2 in the vicinity of H63D.21
In a previous study, we examined 478 probands for C282Y and H63D mutations that accounted for 86.8% and 5.75% of HH chromosomes, respectively.13 Only 7.45% of HH chromosomes did not bear either of these two mutations, and 10.8% of probands were incompletely characterized with at least one chromosome without an assigned mutation. C282Y and H63D substitutions accounted for a total of 92% of HH alleles, suggesting that another mutation(s) is involved in HH. This prompted us to examine the involvement of the S65C substitution in hemochromatosis.
PATIENTS AND METHODS
Probands and control subjects.
The study was performed on a series of 711 unrelated probands with clinical hemochromatosis and 410 unrelated individuals randomly selected as controls. The diagnosis of HH was based on the presence of at least two of the following criteria: a transferrin saturation greater than 60% in males and 50% in females, a serum ferritin concentration greater than 400 μg/L in males and 300 μg/L in females, or a serum iron concentration greater than 20 μmol/L. The amount of iron removed by quantitative phlebotomy was estimated as previously reported.13
Polymerase chain reactions (PCR) and restriction enzyme assay.
DNA was extracted from peripheral blood leukocytes. C282Y and H63D substitutions were analyzed as previously described.13 The 193A→T (S65C) substitution was detected with a HinfI digest, the substitution abolishing a restriction site. First, exon 2 of the HFE gene was partly amplified using the primers and PCR conditions previously described for H63D analysis.13 Then, 10 μL of the PCR products was digested with 5 U of HinfI for 2 hours at 37°C and resolved on a 3% Nusieve 3:1 agarose gel (FMC BioProducts, Rockland, ME). Amplification resulted in a PCR product of 208 bp; the wild-type digest resulted in fragments of 147 bp and 61 bp.
Sequencing.
After specific amplification by PCR, the six coding exons and exon-intron junctions of the HFE gene were all sequenced by dideoxynucleotide chain termination method using a Thermo Sequenase Terminator Cycle Sequencing kit (Amersham Pharmacia Biotech, Uppsala, Sweden).
Statistical analysis.
The Student’s t-test was used to compare the mean value of serum iron, serum ferritin, and transferrin saturation between groups of probands. Fisher’s exact test was used to compare the prevalence of the S65C substitution among HH probands with controls.
RESULTS
Analysis of C282Y and H63D mutations.
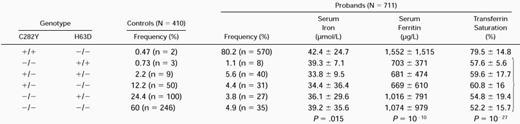
The two substitutions known to be involved in hemochromatosis, namely C282Y and H63D, were analyzed by PCR amplification of exons 4 and 2 of the HFE gene followed by restriction digestion. Seven hundred eleven unrelated probands and 410 control subjects from Brittany were genotyped for C282Y and H63D (Table 1). We detected the C282Y mutation on 85% and 7.7% of patient and control chromosomes, respectively. The C282Y homozygous genotype accounted for 80% of probands and only 0.5% of controls. The H63D variant was present in 5.8% of proband and 14% of control chromosomes and 8 patients (1.1%) and 3 controls (0.7%) were homozygous for this variation, whereas 40 patients and 9 controls were compound heterozygotes C282Y/H63D. The H63D represented 39% of the HH chromosomes that did not carry the C282Y mutation, showing a 2.6-fold increase compared with controls (15%). Among the probands that were neither homozygous nor compound heterozygous, the C282Y heterozygotes accounted for 33%, showing a 2.6-fold increase compared with controls. There was a clear correlation between proband genotype and iron overload. The nonhomozygous probands for C282Y had milder overload compared with the C282Y homozygotes, as shown by initial quantitative phlebotomy (<5 g of iron removed), serum iron, serum ferritin, and transferrin saturation levels at diagnosis.
Analysis of the S65C substitution.
We developed a PCR restriction method to study the 193A→T substitution in the second exon of the HFE gene, which leads to serine 65-to-cysteine substitution. Sixteen S65C substitutions were identified among the 410 control subjects and 10 were observed among the 711 studied probands (Table 2). The S65C variant was never found in individuals with either the homozygous genotypes C282Y/C282Y and H63D/H63D or the compound heterozygotes C282Y/H63D, indicating that the S65C substitution and the other known mutations, C282Y and H63D, are mutually exclusive. Taking into account the control chromosomes that were already known to carry C282Y or H63D (N = 178), a total of 642 control chromosomes carried neither the C282Y substitution nor the H63D, whereas 2.49% of them carried the S65C variant (16/642). Ten probands carried the S65C allele and all of them carried at least one chromosome with an unassigned mutation for HH. Thus, 10.7% (10/93) of HH probands with incompletely characterized genotypes for HH carried the S65C substitution; among them, compound heterozygotes accounted for 16% and 7.4% of C282Y and H63D carriers, respectively. The S65C substitution represented 7.8% (10/128) of the HH chromosomes that were neither C282Y nor H63D, whereas only 2.49% of control chromosomes carried the S65C substitution. This represented a 3.1-fold enrichment compared with controls. Fisher’s exact test was used to compare the frequency of S65C among HH chromosomes with controls. A two-sided exact test provided a P value of .0078. The S65C substitution was thus significantly higher among the HH chromosomes not carrying the C282Y or H63D subtitutions than in controls. We further analyzed theHFE coding sequence of S65C carriers that did not show any additional variations. These 10 hemochromatic patients with the S65C substitution had clinical and biochemical features typical of genetic hemochromatosis (serum iron, 31.2 ± 2.3 μmol/L; serum ferritin, 646 ± 128 μg/L; transferrin saturation, 71% ± 21%).
The C282Y homozygotes and nonhomozygotes constitute two groups of patients associated with severe and mild iron overload, respectively. Analysis of the sex ratio between the two defined groups of HH probands showed that the ratio of male-to-female HH probands was 4:1 (81% of males), whereas among homozygous C282Y and nonhomozygous C282Y, it was 3:1 (72% of males) and 7:1 (90% of males), respectively. Thus, the presence of a mutation with mild effect (H63D, S65C, or other unknown) would increase the risk of developing this disease in males.
DISCUSSION
We have performed an investigation on a large series of HH probands and controls by analyzing two known substitutions involved in HH as well as another substitution, initially described as a polymorphism, occuring in the coding sequence of HFE. Allele frequencies of C282Y, H63D, and S65C were 7.7%, 14%, and 1.95% of control chromosomes, respectively, and 85%, 5.8%, and 0.7% of HH chromosomes, respectively. Moreover, a 2.6-fold increase of the H63D substitution among non-C282Y HH chromosomes and a 2.6-fold increase of the S65C substitution among non-C282Y, non-H63D HH chromosomes compared with controls were significant.
This study confirms previous results concerning the prevalence of the C282Y and H63D substitutions in HFE. Allele frequencies for C282Y and H63D substitutions observed in a control population from Brittany were similar to those reported from a British-Irish population, ie, 6.4% for C282Y and 12.8% for H63D.22 Thus, our results agree with a Celtic origin of the Brittany population. The 845G→A (C282Y) substitution is the most prevalent mutation involved in HH and homozygosity representing 80% of HH probands is associated with the most severe form of this disease, which correlates with the results obtained in vitro showing that the mutation abolishes the HFE function in iron uptake. The HH chromosomes that do not carry the C282Y mutation show a higher frequency of the 187C→G substitution (H63D; 39%) than controls, although only 1.1% of the probands were homozygous. Thus, the H63D substitution is clearly associated with an increased storage of iron, although this variant is associated with a milder form of the disease and shows an incomplete penetrance. This is relevant, because a H63D substitution, at least in vitro, leads only to a reduced activity of HFE protein. In the present study homozygous probands represented 81.1%, compound heterozygotes represented 5.6%, and 62% of the remaining probands were heterozygotes for the C282Y or H63D mutations.
The C282Y and H63D substitutions accounted for a total of 90.8% of HH alleles in our study. The remaining chromosomes showed heterogeneity of haplotypes defined by markers3 13; thus, we might expect to find only rare mutations in the cases in which HH is linked to theHFE gene in these individuals. We examined the prevalence of the 193A→T (S65C) substitution occuring in the coding sequence of HFE in HH probands and controls. Interestingly, 7.2% of HH chromosomes carrying neither the C282Y nor H63D mutations bore the S65C substitution, compared with only 2.49% of control chromosomes. Despite the low frequency of the S65C subtitution, a Fisher exact test showed a statistically significant difference. Among the series of probands, 16% of C282Y heterozygotes and 7.4% of H63D heterozygotes also carried the S65C substitution. Thus, the S65C could be another variant contributing to iron overload in mildly affected hemochromatosis subjects.
Another interesting point is that the male-to-female sex-ratio was 3:1 among the homozygous HH probands for C282Y and 7:1 for the remaining probands. Male individuals carrying two of these mild mutations or one in combination with C282Y would be more at risk of developing hemochromatosis than females. Indeed, due to menstruation and pregnancy, females seem to be protected from developing iron overload,23 which explains why male individuals carrying mild mutations are more prevalent among HH probands than females. Thus, the risk of developing the disease is associated with the HFEgenotype and the subject’s sex.
The functional significance of S65C substitution in mild iron overload should be examined. One can hypothesize that the S65C substitution located in the proximity of H63D may not be able to reduce the affinity of TfR for transferrin as efficiently as the wild-type, as demonstrated for the H63D mutant protein.7 Another possible explanation may be that the 193A→T is a polymorphism linked to a different genetic alteration affecting the level of HFE expression and that S65C has no effect on its own.
In conclusion, our present data provide evidence that the S65C substitution is significantly higher among HH chromosomes that are noncarriers of the C282Y or H63D substitutions. These results bring additional refinement to the correlation between phenotypic heterogeneity and genotype among HH probands. The main mutation associated with high iron overload, C282Y, accounted for 85% of HH chromosomes, whereas the remaining chromosomes were composed mainly of the two variants associated with milder iron overload, H63D and S65C, accounted for 39% and 4.7%, respectively. Homozygotes for C282Y and H63D represented 80% and 1.1%, respectively; the compound heterozygotes consisted of 5.6% of C282Y/H63D and 1% of C282Y/S65C and H63D/S65C.
Supported by grants from CRI 9607 and Association de Transfusion Sanguine et de Biogénétique Gaetan Saleun.
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. section 1734 solely to indicate this fact.
REFERENCES
Author notes
Address reprint requests to Catherine Mura, PhD, Centre de Biogénétique, ETSBO, CHU, UBO, BP454, 46 rue Félix Le Dantec, F-29275 Brest, France; e-mail: Catherine.Mura@univ-brest.fr.