

In this issue of Blood, Zhang et al1 present the largest prospective study reported to date demonstrating the clinical benefit of a first-line targeted therapy for children with hemophagocytic lymphohistiocytosis (HLH). HLH is a rare, life-threatening systemic illness that is characterized by unrestrained T-cell activation and cytokine-mediated hyperinflammation, referred to as a cytokine storm. Clinicians are under enormous pressure to act quickly and decisively to interrupt the cycle of immune activation and tissue destruction when faced with a critically ill child with suspected HLH. Although high-dose glucocorticoids coupled with chemotherapy are commonly used, this approach is associated with significant risks. Ruxolitinib has emerged as a promising treatment option in HLH because of its favorable toxicity profile. The study of Zhang and colleagues provides strong support for ruxolitinib as front-line treatment of HLH and represents a major step forward in updating the approach to this life-threatening disease.
The Histiocyte Society’s HLH 1994 and 2004 clinical trials form the basis for treatment of HLH in children. These studies were designed primarily to improve outcome in patients with inherited defects in lymphocyte cytotoxicity, that is, familial HLH (FHL), which was uniformly fatal prior to their development.
Initial therapy consists of etoposide and dexamethasone to suppress inflammation by reducing T-cell activation, followed by allogeneic hematopoietic cell transplant for patients with genetically verified FHL or refractory/recurrent disease. This strategy results in long-term survival in 61% of cases. Unfortunately, the treatment is both profoundly myelosuppressive and broadly immunosuppressive and associated with pretransplant mortality in 20%.2 Importantly, the need for chemotherapy-based treatments for patients who do not have FHL, that is, those with various forms of secondary HLH, is not established.
In recent years, the development of more-targeted, less-toxic therapy for HLH has emerged as a subject of intense interest and study. Although T-cell activation is important in the pathophysiology of HLH, elevated levels of inflammatory cytokines, such as interleukin-1β (IL-1β), IL-2, IL-6, IL-18, tumor necrosis factor (TNF), and interferon-γ (IFN-γ), are increasingly recognized as major drivers of disease activity. The importance of neutralizing inflammatory cytokines was first noted in macrophage activation syndrome (MAS), a form of secondary HLH, where blockade of IL-1β with anakinra, an IL-1 receptor antagonist, is highly effective.3 More recently, neutralization of IFN-γ with emapalumab led to its Food and Drug Administration approval for treatment of FHL in patients resistant to or intolerant of standard chemoimmunotherapy.4
Ruxolitinib, an inhibitor of Janus kinase 1 (JAK1) and JAK2, blocks the signaling of cytokines through JAK/STAT (signal transducer and activator of transcription) pathways, endowing it with the ability to simultaneously inhibit the action of IFN-γ, IL-2, IL-6, and other proinflammatory cytokines (see figure). Ruxolitinib has demonstrated activity in mouse models of primary and secondary HLH.5 Case series of adults with refractory HLH provide evidence that ruxolitinib can be effective as salvage therapy. However, there are few reports on the use of this medication as front-line therapy in adults or children with HLH.6-8
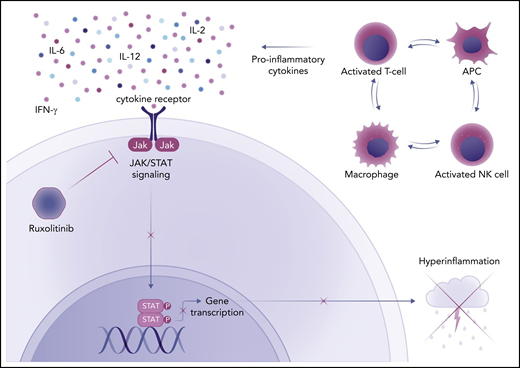
The mechanism of action of ruxolitinib in HLH. Activated T cells, natural killer (NK) cells, macrophages, and antigen-presenting cells (APC) produce proinflammatory cytokines in HLH. Many of these cytokines (IFN-γ, IL-2, IL-6, IL-12) signal through the JAK/STAT pathway to induce changes in gene expression that lead to hyperinflammation and cytokine storm. Ruxolitinib, a JAK1/2 inhibitor, blocks this process. Professional illustration by Somersault18:24.
The mechanism of action of ruxolitinib in HLH. Activated T cells, natural killer (NK) cells, macrophages, and antigen-presenting cells (APC) produce proinflammatory cytokines in HLH. Many of these cytokines (IFN-γ, IL-2, IL-6, IL-12) signal through the JAK/STAT pathway to induce changes in gene expression that lead to hyperinflammation and cytokine storm. Ruxolitinib, a JAK1/2 inhibitor, blocks this process. Professional illustration by Somersault18:24.
Zhang et al tested the efficacy of ruxolitinib monotherapy in this single-center, single-arm prospective study of 52 children who fulfilled standard diagnostic criteria for HLH. Cotreatment with glucocorticoids was permitted. Patients were stratified by their initial response to the JAK inhibitor, and those with an unfavorable response received individualized intensification treatment with chemotherapy (mostly etoposide and methylprednisolone ± other agents) in addition to ruxolitinib. The primary endpoint was the overall response rate to ruxolitinib monotherapy; this was achieved in 69% of patients. A complete response to ruxolitinib monotherapy was observed in 42% of patients, all of whom demonstrated response by day 3, completed 28 days of ruxolitinib, and subsequently remained disease free. Of the 58% of patients who required addition of “intensive” chemotherapy, about half had treatment response. Ruxolitinib monotherapy followed by intensification therapy in poor responders resulted in 83% overall survival. Among the 52 patients, HLH was associated with Epstein-Barr virus (EBV) in 34 cases (65%). The patients with EBV-HLH were significantly more likely, whereas those with chronic active EBV-HLH were less likely to survive.
There are several important findings to highlight. The study provides strong evidence that a rapid and durable response to ruxolitinib alone can be achieved in a significant subset of children with HLH, obviating their need for toxic chemotherapy. The response-stratified addition of chemotherapy to ruxolitinib appears to be safe with no obvious downside. However, treatment must be rapidly intensified in patients who do not respond within the first few days, as all partial and nonresponders in this study progressed. In stark contrast to standard chemoimmunotherapy, ruxolitinib was exceptionally well tolerated with few attributable serious adverse events.
There are caveats to consider. The study was conducted at a single center, and the majority of the enrolled patients were diagnosed with secondary HLH. This study cohort may not reflect the full range or distribution of HLH cases seen in all hospitals or populations. In particular, the high proportion of EBV-related cases is noteworthy. In addition, the exclusion of suspected/confirmed FHL and MAS prevents insights into the potential utility of ruxolitinib for these patients. Whether ruxolitinib will be efficacious in patients with severe illness and/or multisystem organ failure remains to be determined, as these patients were not included in the study.
The results of this study are encouraging and will advance pediatric practice. However, clearly more work needs to be done to confirm the findings in other settings, to define the optimal dose of ruxolitinib, and to identify the patient population(s) most likely to benefit from it. The results underscore the heterogeneity of HLH, which confounds easy, one-size-fits-all solutions. Better diagnostic tools and biomarkers are needed to help clinicians identify the subset of children with HLH who will respond well to ruxolitinib and/or other targeted agents. When ruxolitinib fails, the best next steps, that is, cytotoxic chemotherapy, immunosuppressants, or other targeted treatments, need to be determined through high-quality comparative study. Although there is still much to learn, we are getting smarter.
Conflict-of-interest disclosure: L.A.H. received salary support from the Childhood Arthritis and Rheumatology Research Alliance and consulting fees from Sobi, Pfizer, and Adaptive Biotechnologies. None of these are relevant to this manuscript. B.A.D. declares no competing financial interests.

