Abstract
Humans produce and remove 1011 platelets daily to maintain a steady-state platelet count. The tight regulation of platelet production and removal from the blood circulation prevents anomalies in both processes from resulting in reduced or increased platelet count, often associated with the risk of bleeding or overt thrombus formation, respectively. This review focuses on the role of glycans, also known as carbohydrates or oligosaccharides, including N- and O-glycans, proteoglycans, and glycosaminoglycans, in human and mouse platelet and megakaryocyte physiology. Based on recent clinical observations and mouse models, we focused on the pathologic aspects of glycan biosynthesis and degradation and their effects on platelet numbers and megakaryocyte function.
Introduction
Platelets circulate in blood at an average range of 150 to 450 × 109/L in adults. Platelets are small (1-2 μm) enucleated cells produced by megakaryocytes. Their principal function is to prevent bleeding, but platelets are functionally diverse and contribute to immune responses and metastatic processes.
Platelet production and clearance are tightly regulated; an imbalance in either process can lead to detrimental complications, including thrombus formation. This review focuses on the role of glycans, also called carbohydrates or oligosaccharides, including N- and O-glycans, proteoglycans, and glycosaminoglycans (GAGs), in human and mouse platelet and megakaryocyte physiology. The review addresses pathologic aspects of altered glycosylation with regard to platelet numbers and megakaryocyte function.
Glycosylation overview
Glycobiology studies the structure, biosynthesis, biology, and evolution of glycans. Glycans are present on the surface of every cell as a significant component of a sugar coating termed glycocalyx. Glycans are also major constituents of extracellular spaces, including the bone marrow extracellular matrix (ECM) and blood lining. Adding glycans to a cell or macromolecule, such as a protein or lipid, is a complex multistep process involving >200 enzymes composed of glycosyltransferases and glycosidases. Unlike protein synthesis, glycosylation is a nontemplate process. A particular glycan structure (ie, its length and position) cannot be simply predicted by gene expression; instead, it is dictated by the expression and kinetic properties of enzymes and the sugar acceptors involved. Glycoenzyme expression, however, is gene expression profile dependent, cell specific, and sensitive to the physiologic cell state dictated by environmental cues. These properties define individual glycome cell, organ, and individual profiles.
Because of their hydrophilic nature, glycans are mostly cell surface constituents among proteins and lipids. Glycans and glycan-binding receptors (lectins) regulate cell-cell interactions, cell adhesion, migration, immune responses, and self/nonself-recognition, best exemplified by the ABO blood group system. Glycans, classified according to the glycosidic bond as N or O linked, add to biologic complexity by cell-specifically modifying the biologic properties of specific proteins and lipids. N- and O-glycosylation are posttranslational modifications that occur after the protein has been synthesized in the endoplasmic reticulum and Golgi apparatus. Given the extent of the glycan literature, we will focus this review on glycans relevant to megakaryocytes and platelets.
N-glycosylation
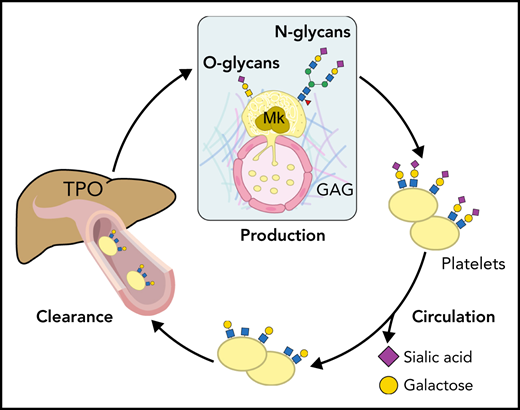
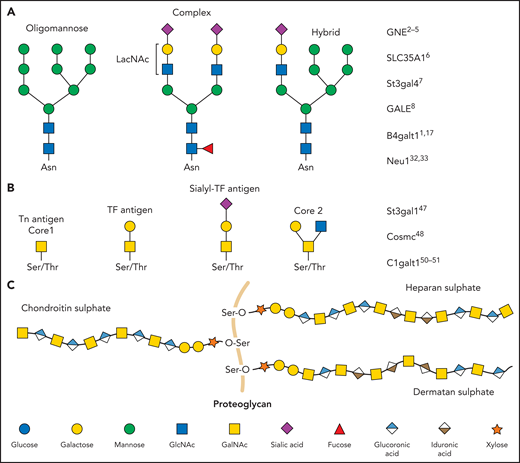
N-linked glycans (ie, N-glycans) are bound to proteins at asparagine (Asn) residues by an N-glycosidic bond. In eukaryotes, N-glycans begin with N-acetylglucosamine (GlcNAc) β1-Asn and share the typical core sequence, mannose (Man) α1-3(Manα1-6)Manβ1-4GlcNAcβ1–4GlcNAcβ1–Asn-X-serine (Ser)/threonine (Thr). Three types of N-glycans exist based on core extensions: (1) oligomannoses, where only Man moieties are added; (2) complex N-glycans with antennae initiated by GlcNAc additions; and (3) hybrids, where the Manα1-6 arm extends the core and 1 or 2 GlcNAcs extend the Manα1-3 arm (Figure 1). The core structures in Figure 1 show terminal GlcNAc and may be extended by β1-4galactose (Gal) to generate a type-2 unit composed of Galβ1-4GlcNAc (LacNAc). Poly-LacNAc (-3Galβ1-4GlcNAcβ1-)n is generated by the addition of β1-3GlcNAc and further elongation by β1-4Gal, thus forming 2 LacNAc units. LacNAc decorations, specifically sialic acid (Sia), determine platelet lifespan and play a role in thrombopoiesis.1,2
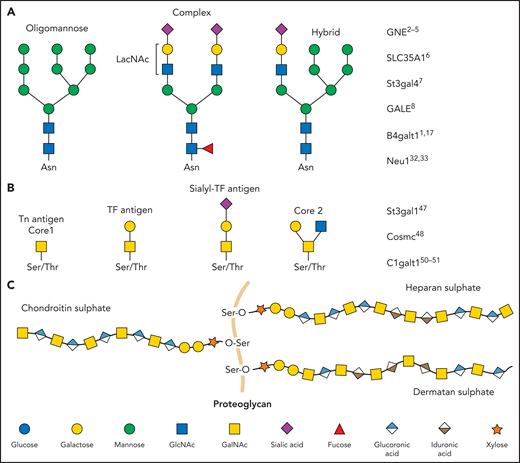
Glycan diversity. Schematic figures of glycans and genes that regulate platelet production and clearance. (A) N-glycans are bound to glycoproteins through an N-glycosidic bond to asparagine (Asn). Depending on how their core is branched out, they can be classified as oligomannose, complex, or hybrid. (B) O-glycans are bound through an O-glycosidic bond to serine (Ser) or threonine (Thr). (C) Proteoglycans consist of a protein core (orange) and ≥1 covalently attached GAG chains. GAGs consist of repeating disaccharide units composed of an N-acetylated or N-sulfated hexosamine and either a uronic acid (GlcA or IdoA) or galactose. LacNAc, N-acetyllactosamine; TF, Thomsen-Friedenreich.
Glycan diversity. Schematic figures of glycans and genes that regulate platelet production and clearance. (A) N-glycans are bound to glycoproteins through an N-glycosidic bond to asparagine (Asn). Depending on how their core is branched out, they can be classified as oligomannose, complex, or hybrid. (B) O-glycans are bound through an O-glycosidic bond to serine (Ser) or threonine (Thr). (C) Proteoglycans consist of a protein core (orange) and ≥1 covalently attached GAG chains. GAGs consist of repeating disaccharide units composed of an N-acetylated or N-sulfated hexosamine and either a uronic acid (GlcA or IdoA) or galactose. LacNAc, N-acetyllactosamine; TF, Thomsen-Friedenreich.
Cell-specific expression, regulation of glycosyltransferase activities, and availability of critical acceptor/donor substrates dictate the structural diversity of glycoprotein N-glycan modifications. N-glycan diversification is used by cells as control checkpoints to establish tissue- and cell-specific unique glycan expression patterns during developmental stages in response to cell surroundings. The N-glycan checkpoints are necessary for the formation and creation of functional tissue.3 Glycosylation-mediated quality control of protein folding by N-glycans is another significant task of N-glycosylation. Man-6-phosphate N-glycan determinant recognition facilitates quality control by targeting misfolded proteins to lysosomes.4
Sialylation creates substantial glycan diversity. Sialyltransferases (STs) catalyze the linkage of the C-2 Sia carbon to galactose in C-3 or C-6 or N-acetylgalactosamine (GalNAc) C-6 positions, which results in α-2,3 or α-2,6 linkages, respectively. STs differential and organ-specific expression further diversifies the functions of Sias.5 Sialylated derivatives of the glycan structure β4-LacNAc (Galβ1,4GlcNAc or type-2 LacNAc; hereafter referred to as LacNAc) regulate platelet lifespan, hepatic thrombopoietin (TPO) production, and thrombopoiesis.6 However, the detailed mechanisms if perturbed glycosylation affects platelet production or clearance are not always precisely defined.
Role of megakaryocyte-intrinsic glycans in platelet production and platelet clearance
Genetic disorders affecting platelet sialylation and galactosylation in humans and different mouse models have been studied extensively. Congenital thrombocytopenia in humans is associated with mutations in the GNE gene encoding the UDP-GlcNAc 2-epimerase/ManNAc kinase, an enzyme that controls Sia synthesis,7-10 and in the Sia transporter gene SLC35A1.11GNE and SLC35A1 mutations likely lead to increased desialylated clearance, causing thrombocytopenia. Mouse models such as St3gal4-knockout mice lacing α-2,3-ST 4 (ST3Gal4) have profound thrombocytopenia. Crossing St3gal4-null mice with mice lacking the hepatic Ashwell-Morell receptor (AMR), also called the asialoglycoprotein receptor, results in platelet count recovery, implying that Gal uncovering leads to AMR-mediated clearance.12 Whether AMR or other Gal-recognizing hepatic lectins mediate platelet clearance in patients with GNE and SLC35A1 mutations requires further investigation.
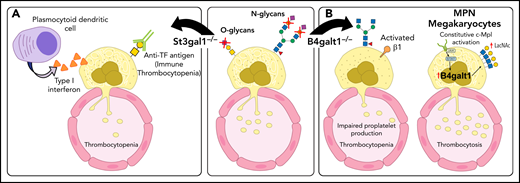
GALE encodes the UDP-galactose-4-epimerase, an enzyme essential to Gal metabolism. Rare GALE mutations (p.R51W [c.C151T, NM_001127621]) cause severe thrombocytopenia with dysplastic megakaryocytes, suggesting a defect in thrombopoiesis.13 Deletion of the β-1,4-galactosyltransferase 1 (β4GalT1) in mice results in profound thrombocytopenia with dysplastic megakaryocytes with abnormal demarcation membrane systems and hyperactive β1 integrin, leading to defective thrombopoiesis and thrombocytopenia (Figure 2).6
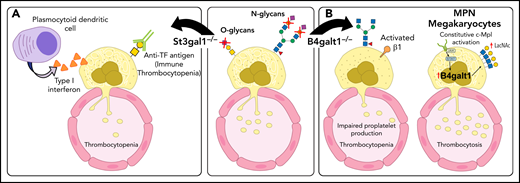
Glycosylation changes affect platelet production directly and indirectly. (A) Deficit in the sialyltransferase ST3Gal1 (St3gal1MK−/−) causes exposure of the cryptic TF antigen that is recognized by a plasmocytoid dendritic-like cell inducing the release of type 1 interferon dampening platelet production. Increased immunoglobulin G antibodies against the TF antigen are observed in pediatric patients with immune thrombocytopenia (ITP). (B) β4GalT1 regulates platelet production. Megakaryocytes derived from B4galt1−/− mice have an underdeveloped demarcation membrane system, β1 integrin hyperactivity, and impaired platelet production, causing thrombocytopenia. Increased expression and activity of β4GalT1 is observed in megakaryocytes isolated from patients with myeloproliferative neoplasms (MPNs) with high allele burden. Highly galactosylated platelets in MPNs may promote increased hepatic TPO synthesis that sustains the aberrant megakaryopoiesis. IL-6, interleukin 6.
Glycosylation changes affect platelet production directly and indirectly. (A) Deficit in the sialyltransferase ST3Gal1 (St3gal1MK−/−) causes exposure of the cryptic TF antigen that is recognized by a plasmocytoid dendritic-like cell inducing the release of type 1 interferon dampening platelet production. Increased immunoglobulin G antibodies against the TF antigen are observed in pediatric patients with immune thrombocytopenia (ITP). (B) β4GalT1 regulates platelet production. Megakaryocytes derived from B4galt1−/− mice have an underdeveloped demarcation membrane system, β1 integrin hyperactivity, and impaired platelet production, causing thrombocytopenia. Increased expression and activity of β4GalT1 is observed in megakaryocytes isolated from patients with myeloproliferative neoplasms (MPNs) with high allele burden. Highly galactosylated platelets in MPNs may promote increased hepatic TPO synthesis that sustains the aberrant megakaryopoiesis. IL-6, interleukin 6.
Acquired driver mutations in hematopoietic stem cells cause MPNs. These mutations include genes encoding the tyrosine kinase JAK2 (JAK2),14-17 the endoplasmic reticulum protein calreticulin (CALR),18,19 and the TPO receptor MPL (MPL).20,21 Plasma TPO levels in MPNs do not correlate with the platelet or megakaryocyte mass. Patients with MPNs with a high allele burden of the mutated clones have increased TPO plasma, despite their increased platelet count. Megakaryocytes derived in vitro from patients with MPNs showed an increased expression of the B4GALT1 gene encoding β4GalT1. Consistently, megakaryocytes and circulating platelets have increased terminal Gal expression relative to healthy controls, ameliorated by a JAK1/2 inhibitor and correlated with the high allele burden regardless of the underlying identified mutation (Figure 2). We suggest that altered β4GalT1 expression in mutant megakaryocytes and platelets increases terminal Gal levels and clearance via liver AMR, thus promoting TPO production by hepatocytes independent of platelet mass.22 The data reveal that Sia and Gal metabolism are critical for platelet clearance and production in both humans and mice. These data also indicate that impaired intracellular signaling in megakaryocytes can be associated with megakaryocyte and platelet surface glycan perturbations, thereby affecting platelet production and clearance.
Sia and circulatory platelet lifetime
In 1975, Greenberg et al23 used neuraminidase to remove Sia from platelets in vitro, a maneuver that led to untimely platelet destruction after transfusion, implying that Sia loss targets platelets for clearance. Platelets also lose Sia during storage in vitro24 and upon aging in vivo.12,25 Studies from independent laboratories have found that liver hepatocytes remove desialylated platelets using their AMR.12,26,27 However, the discussion regarding the function of hepatocyte lectins in platelet removal persists.
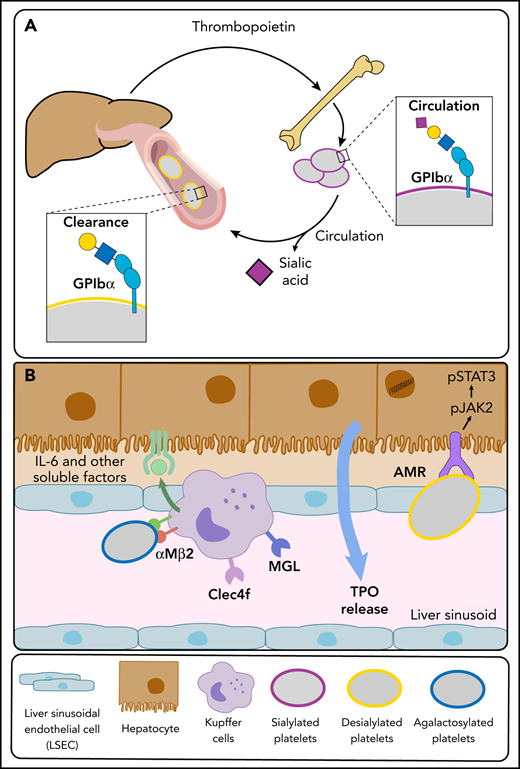
The AMR C-type lectin is highly conserved and recognizes uncovered β-Gal and GalNAc, generally covered by Sia. Different subunits (ASGR) combine as homo- and hetero-oligomers to form an array of AMR conformations, likely altering substrate selectivity and binding affinities.28 The ASGR-12/ASGR-2 trimer core is the most abundant and has the highest affinity.29,30 Mice lacking ASGR-1 or ASGR-2 lack functional hepatic AMRs that recognize platelets lacking Sia (Figure 3),12,31,32 as demonstrated by the following findings: Asgr2-knockout mice have increased circulating desialylated platelets with a prolonged circulatory lifespan, an expected result if AMR routinely removes desialylated senile platelets, and in vitro or in situ platelet desialylation by neuraminidases causes rapid platelet clearance in wild-type mice but not in Asgr2-null mice, independent of macrophage depletion.12 Subsequently, additional macrophage (Kupffer cell) lectins, including macrophage Gal lectin,27 integrin αMβ2,33 and Clec4f,34 were found to identify desyalylated and degalactosylated platelets, giving further credence to the notion that glycan-mediated platelet clearance is more complex than anticipated (Figure 3).
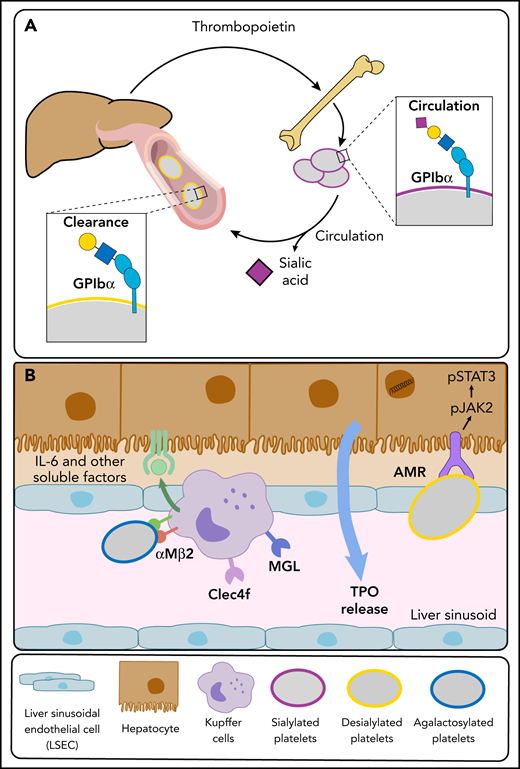
Platelet and liver TPO production feedback loop. (A) At steady state, completely sialylated platelets (purple circles) circulate through the hepatic sinusoids. Aged platelets with heightened terminal Gal and GalNAc residues (yellow circles) are cleared in the liver, initiating hepatic TPO production. (B) The hepatic AMR directly recognizes desialylated platelets, activating the JAK2-STAT3 pathway to elicit TPO production. Various lectins on liver macrophages (Kupffer cells), including the integrin αMβ2, the macrophage Gal lectin (MGL), and Clec4f bind desialylated platelets. Platelet binding to macrophage lectins could trigger cytokine release, including IL-6, and induce IL-6 receptor (IL-6R) responses to stimulate TPO production further.
Platelet and liver TPO production feedback loop. (A) At steady state, completely sialylated platelets (purple circles) circulate through the hepatic sinusoids. Aged platelets with heightened terminal Gal and GalNAc residues (yellow circles) are cleared in the liver, initiating hepatic TPO production. (B) The hepatic AMR directly recognizes desialylated platelets, activating the JAK2-STAT3 pathway to elicit TPO production. Various lectins on liver macrophages (Kupffer cells), including the integrin αMβ2, the macrophage Gal lectin (MGL), and Clec4f bind desialylated platelets. Platelet binding to macrophage lectins could trigger cytokine release, including IL-6, and induce IL-6 receptor (IL-6R) responses to stimulate TPO production further.
Desialylation was reported in aging red blood cells (RBCs) and β-thalassemia ∼40 years ago.35,36 Although desialylated RBCs are rapidly cleared and sequestered by the liver, it is unclear whether the mechanisms involved are like those affecting platelets. Interestingly, young but not old RBCs or platelets can repair desialylation, consistent with different behaviors in vivo.36
Platelets express neuraminidase Neu1, Neu2, and Neu4. The addition of anti-GPIbα antibodies is associated with increased surface Neu1 in a subset of patients with ITP.37,38 Influenza antiviral drugs, including zanamivir (Relenza), oseltamivir (Tamiflu), and peramivir (Rapivab), mimic the transition state to block viral neuraminidase. Administration of the neuraminidase inhibitor oseltamivir increases platelet counts in some patients with ITP,39-41 as well as in healthy individuals, suggesting that platelet Sia regulates platelet counts.42 Thus, some patients with ITP with desialylated platelets could profit from sialidase inhibitor administration or perhaps from modification of the function of Sia-binding immunoglobulin-like lectins (Siglecs).
Desialylated platelets and the liver and bone marrow feedback loop
The enigma of how platelets interact with hepatic AMR remains unsolved. Clear evidence from several groups supports the notion that AMR recognizes desialylated platelets, an interaction facilitated by desialylated GPIbα, a von Willebrand factor receptor subunit.12,31,32 This interaction is thought to elicit hepatic TPO production.43,44 The clearance mechanism via AMR might include >1 receptor. In recent work, Voos et al45 showed that exogenous neuraminidases, enzymes that remove Sia, cause desialylation of O-glycans in the subunit GPIbα, inducing the unfolding of the mechanosensory domain triggering GPIb-IX signaling. This activation causes degranulation and exposure of endogenous Neu1, exposing galactose on N-glycans, which is finally recognized by AMR.
Furthermore, a role of GPIbα-bound von Willebrand factor associated with asialoglycans cannot be excluded. The significance of the hepatocyte-platelet interchange rises past simple removal. Platelet removal by hepatic AMR causes JAK-STAT signaling events that affect TPO production, the integral controller of megakaryopoiesis.12 Recent data show that the IL-6R also regulates platelet count and hepatic TPO production at a steady state,26 as we suggested earlier. The following intriguing data support the role of IL-6R in TPO and platelet number control: IL-6R–null mice have increased hepatic AMR expression and higher platelet counts, with larger and increased numbers of reticulated (RNA-containing) platelets. These data imply younger newly produced platelets supported by fewer circulating platelets with terminal galactose, suggesting an increased desialylated platelet clearance leading to the measured high hepatic TPO messenger RNA levels.26 Therefore, IL-6R and AMR synergistically regulate TPO production in the liver.
O-glycosylation
O-linked glycosylation is the attachment of a sugar molecule to Ser or Thr residues in a protein. O-glycan decorations and structures affect protein stability and regulate protein activity. O-glycans have numerous functions throughout the body, including trafficking cells in the immune system, recognizing foreign material, and controlling cell metabolism.46 Because of their many functions, changes in O-glycans are essential in many diseases, including inflammation, cancer, diabetes, Alzheimer’s, and thrombopoiesis. O-glycan biosynthesis is initiated by GalNAc addition onto a Ser or Thr residue from a precursor molecule through the activity of a GalNAc transferase enzyme. Unlike N-glycans, the specific residue onto which GalNAc will be added is not well defined, because numerous enzymes can add the sugar, and each will favor different residues.
Two of the most common O-glycan structures formed are core 1 and core 2 (Figure 1). O-GalNAc sugars are essential in leukocyte trafficking and platelet function.47 P-selectin glycoprotein ligand-1 is such a ligand, and it contains various O-glycan structures necessary to function.48-50 Mucins are heavily O-glycosylated proteins lining the gastrointestinal and respiratory tracts to protect these regions from infection. The negative charge of mucins allows them to interact with water, prevent it from evaporating, regulate inflammation, and protect against infections.51 O-glycans are also essential for the synthesis of the different blood groups. The addition of fucose sugars by fucosyltransferases forms Lewis epitopes and the foundation for blood group determinants. The addition of fucose alone creates the H-antigen, present in individuals with blood group O. Adding a GalNAc sugar will make the A-antigen for blood group A. Alternatively, adding a Gal onto this structure generates the B-antigen for blood group B.52
O-glycans and platelet counts
Multiple changes in O-glycans can also produce severe thrombocytopenia, including deficits in α-2,3-sialyltransferase 1 (ST3Gal1),53 N-acetylgalactosaminyltransferase 3 (GalNT3), and core 1 synthase (C1GalT1) or its chaperone, Cosmc.54-57 ST3Gal1 adds Sia specifically on the core-1 O-glycans, also termed the TF antigen. A mouse model with targeted deletion of St3gal1 in megakaryocytes (St3gal1MK−/−) using the Pf4-Cre mouse model revealed that: (1) TF antigen exposure is restricted to megakaryocytes and does affect other bone marrow cells, resulting in moderate thrombocytopenia in St3gal1MK−/− mice; (2) deletion of the lymphoid-specific tyrosine kinase JAK3 in St3gal1MK−/− mice normalizes platelet counts, thereby implicating the involvement of immune cells in causing thrombocytopenia; and (3) interferon-producing Siglec H+ bone marrow immune cells engage with O-glycan Sia moieties to regulate type 1 interferon secretion and thrombopoiesis, likely facilitated by a new population of immune cells with a plasmacytoid dendritic cell–like signature and concomitant upregulation of immunoglobulin rearrangement gene transcripts Igkc and Ighm, suggesting additional immune regulatory mechanisms.53 Thus, aberrant TF antigen moieties, often found in pathologic conditions, regulate immune cells and thrombopoiesis in the bone marrow, reducing platelet counts. The data show that Sia moieties and decorations have a variety of functions in megakaryocytes and platelets. Siglecs regulate immune responses, often playing an immune inhibitory function.58 Signaling in leukocytes is also regulated by Siglecs, which recognize Sia-containing ligands as self-associated molecular patterns. However, the role of Siglecs in the bone marrow niche has not been widely explored.
Recently published data using the St3gal1MK−/− mouse model poses the intriguing question of whether Siglecs in the bone marrow niche regulate immune responses dictated by the Sia status of cells, including megakaryocytes, in patients with ITP. ITP is a platelet disorder in pediatric and adult patients. ITP in pediatric and adult patients has been associated with Sia alterations, but the pathophysiology of ITP remains elusive, and ITP is often a diagnosis of exclusion. Lee-Sundlov et al53 analyzed pediatric ITP plasma samples, which showed increased anti–TF antigen antibody representation, suggesting increased exposure of the typically sialylated and cryptic TF antigen in these patients.
GAGs and proteoglycans
Sulfated GAGs, including heparin (HP)/heparan sulfate (HS) and dermatan sulfate/chondroitin sulfate (Figure 1) are natural linear polysaccharides. GAGs remain poorly understood in terms of both structure and function. An ionic bond is formed between electronegative charges on the GAGs (eg, SO3− and COO−) and electropositive regions on these proteins (eg, multiple Arg, Lys, and His residues). Based on genomic analysis, ∼435 human proteins bind to HP/HS.59-63 This forms the theoretic HP/HS interactome, which includes: (1) ECM proteins, such as metalloproteases, heparanase, growth factors, and chemokines; (2) intracellular proteins, such as neutrophil elastase, pTAU, and others; (3) cell surface receptors, such as G6b-B, FGF-Rs, VEGF-R, RAGE, and NRPs; (4) nuclear proteins, such as p300, pCAF, and HMBG1; and (5) plasma proteins, such as coagulation proteases, complement factors, morphogens, serpins, and others.
GAGs modulate a myriad of cell processes, including signaling, growth, proliferation, adhesion, and coagulation, and pathologic processes, such as cancer.64,65 Some of the most characterized GAG-protein interactions include the high-affinity and high-selectivity HP-antithrombin system and the low-affinity HP-thrombin system.66-69 Virtually all mammalian cells produce proteoglycans that can be secreted into the ECM, inserted into the plasma membrane, or stored in granules.70 However, there is little understanding of the expression or role of GAGs in the bone marrow, or of what types of GAG structures are present on megakaryocytes or platelets to regulate their function.
One example where differences in platelet GAG expression may play a role in shaping immune responses is HP-induced thrombocytopenia (HIT), a prothrombotic disorder initiated by antibodies to platelet factor 4 (PF4)/HP complexes. PF4 released from platelets binds to surface GAGs expressed on hematopoietic and vascular cells, including platelets. The cell-specific and heterogeneous GAG composition accounts for the differences in affinity for PF4. The structural differences of GAGs on platelet and megakaryocyte surfaces are largely unknown. It is tempting to speculate that interindividual differences in GAG-platelet expression may contribute to differences in PF4 binding and conformational presentations of the PF4-GAG complexes that promote pathologic antibody production, causing HIT.71,72
Role of GAGs and proteoglycans in platelet production
HS proteoglycans, including syndecans 1 to 4, glypican-1, and perlecan, are constituents of the bone marrow stroma under physiologic conditions.73,74 The role of proteoglycans in hematopoiesis was predicted in 1983 by 2 different groups reporting a correlation between GAG biosynthesis in the stroma and the production of pluripotent stem cells.75,76 Several studies reported that GAGs (HS, HP, dermatan sulfate, chondroitin sulfate, and hyaluronic acid) play a role in megakaryopoiesis.77 The proteolytically released C-terminal fragment of perlecan, called endorepellin, binds to integrin α2β1 and enhances collagen-mediated platelet activation.78 A recent study identified HS as the critical regulator of the immune receptor tyrosine-based inhibition motif–containing receptor G6b-B in platelets and megakaryocytes.79 Loss of G6b-B results in severe macrothrombocytopenia, myelofibrosis, and aberrant platelet function in mice and humans.80 Low molecular weight HP, given prophylactically to prevent veno-occlusive events in the liver in patients undergoing bone marrow transplantation, stimulates megakaryopoiesis, thereby accelerating platelet recovery and concomitantly reducing platelet transfusions in low molecular weight HP–treated patients and mice.81,82 Another study showed that 2-O,3-O-desulfated HP blocks PF4 and ameliorates thrombocytopenia in chemotherapy- or radiation-injured mice.83 Thus, HS GAGs and proteoglycans regulate thrombopoiesis and platelet function in vivo. However, their precise role remains unclear.
Schick et al84 showed that megakaryocytes synthesize and store proteoglycans in α-granules, specifically chondroitin sulfate proteoglycans, which possibly bind the cytokine PF4. Still, the role of megakaryocyte-derived proteoglycans in PF4 regulation awaits more work, including in the context of HIT. The PF4-GAG connection could be particularly intriguing in patients with HP-induced thrombocytopenia. Emerging models of hematopoiesis propose a close spatial and genetic relationship between the megakaryocyte lineage and platelet-biased hematopoietic stem cells,85 a population directly differentiated into megakaryocytes to aid platelet production under stress conditions.86 The idea that megakaryocytes regulate parts of this niche by producing ECM molecules, including GAGs, is intriguing and perhaps warrants investigation.
In 1996, Tajika et al87 showed that GAGs specifically affect mature megakaryocytes, promoting proplatelet formation. The study specifically showed that GAG sulfation, rather than GAG length, is essential for thrombopoiesis. The addition of HS and chondroitin sulfate to culture media containing TPO enhances the differentiation of CD34+ hematopoietic progenitors to megakaryocytes by ∼70%, whereas TPO alone yielded ∼35% enhancement.88-91 The Lanza group showed that adding hirudin and HP to culture media enables efficient megakaryocyte differentiation of mouse bone marrow progenitors when added to culture media.92 Thus, GAGs seem to support megakaryopoiesis and thrombopoiesis in vitro and could provide a novel foundation on which to promote in vitro platelet production.
Cardiovascular disorders and the role of platelet glycans and selectins
Role of selectins in reperfusion injury
Vascular thrombotic events such as stroke and myocardial infarction temporarily interrupt blood flow. Natural or medical interventions that restore blood flow suddenly reintroduce leukocytes to the traumatized tissue and can cause tissue damage called reperfusion injury. Selectins, cell adhesion molecules that are C-type lectins, play a significant role in this process. P-selectin (CD62P) on the activated endothelium and platelets in the reperfused area and L-selectin (CD62L) on leukocytes initiate this cascade, followed by E-selectin (CD62E).93 HP can block P- and L-selectin interactions, but the dampening effects of selectin inhibitors on reperfusion injury remain to be established. Small-molecule inhibitors and biologic agents (eg, monoclonal antibodies) to block these interactions in patients have been generated and approved by the US Food and Drug Administration for clinical indications such as sickle cell vaso-occlusive crises, showing beneficial results.94
Role of selectins and Sia in atherosclerosis
Heart attacks, strokes, and other vascular diseases are associated with decreased high-density lipoprotein cholesterol and high levels of low-density lipoprotein (LDL) cholesterol. Atherosclerotic lesions begin with a fatty streak, likely a result of increased LDL levels. Reduced sialylation of LDL in patients also correlates with coronary artery disease. Although the mechanisms remain unclear, desialylated LDL may be more easily taken up and incorporated into atheromatous plaques.95 Monocytes enter the subendothelial regions of the blood vessels to resolve the accumulation of fatty streaks, a process supported by activated platelets that involve P- and E-selectin upregulation on activated platelets and endothelial cells. P- and/or E-selectin recognizes glycosylated and sulfated P-selectin glycoprotein ligand-1 or sialyl Lewis X (CD15s) displayed on discrete glycoproteins and glycolipids of circulating platelets and monocytes. The notion that P-selectin is a critical molecule in atherosclerosis is supported by data showing that mice lacking P-selectin have delayed progression of atherosclerotic lesions.96 Whether inhibition of P- and L-selectin has any effect on reperfusion atherosclerotic progression remains to be established.
Conclusion
The summarized findings provide new insights into the role of glycans in megakaryopoiesis, thrombopoiesis, platelet lifespan, and function.
- 1.
Alterations of the typical terminating glycan structure, sialyl lactosamine, profoundly affect thrombopoiesis and platelet lifespan. Notably, the galactosyltransferase β4GalT1 that partakes in lactosamine is necessary for thrombopoiesis. However, the lack of β4GalT1 does not affect the platelet lifespan. Sias that decorate lactosamine structures and are synthesized by ST3Gal4 play a substantial role in platelet survival, because platelets lacking ST3Gal4 are cleared rapidly via hepatic AMR. On the other hand, Sia decorations of common O-glycan structures, such as core 1 or the TF antigen synthesized by ST3Gal1, play a role in communicating via immune cells expressing Siglecs to regulate thrombopoiesis.
- 2.
Additional Sia perturbations also function in diseases with altered immune responses, including MPNs and ITP. Many genetic alterations that code for glycosyltransferases or Sia transporters have been identified and support the role of glycan platelet count control, but intricate mechanisms remain to be established.
- 3.
Glycans of the ECM (ie, GAGs and proteoglycans) play a role in platelet production. This insight poses the question of whether specific signals and cytokines, including TPO, depend on GAG expression to regulate megakaryopoiesis and thrombopoiesis.
- 4.
There is a long-standing role for selectins and platelet glycans in cardiovascular disorders, such as reperfusion injury and atherosclerotic lesions.
- 5.
Many questions remain unresolved. What are the mechanisms leading to interactions between hepatocytes and platelets? Are glycoforms essential for thrombopoiesis and cross talk in the bone marrow environment? Do specific Siglecs on immune cells regulate thrombopoiesis? Future discussions and investigations are necessary to answer remaining questions to understand the genesis and death of platelets.
Acknowledgments
This work was supported by National Heart, Lung, and Blood Institute, National Institutes of Health, grants R01 HL126743 (H.F.), R01 HL089224 (K.M.H.), and P01 HL107146 and K12 HL141954 (K.M.H. [project leader]).
Authorship
Contribution: H.F., L.R., and K.M.H. contributed equally to literature review and manuscript writing; and L.R. created the figures.
Conflict-of-interest disclosure: K.M.H. has received consulting fees from Pfizer. The remaining authors declare no competing financial interests.
Correspondence: Karin M. Hoffmeister, Versiti Blood Research Institute, 8733 W Watertown Plank Rd, Milwaukee, WI 53217; e-mail: khoffmeister@versiti.org.