Key Points
TAK-981 enhances macrophage phagocytosis and NK cell cytotoxicity in combination with rituximab via IFN1 pathway activation.
Combined treatment with TAK-981 and rituximab promotes synergistic in vivo antitumor activity in CD20+ lymphoma xenograft models.

Abstract
Small ubiquitin-like modifier (SUMO) is a member of a ubiquitin-like protein superfamily. SUMOylation is a reversible posttranslational modification that has been implicated in the regulation of various cellular processes including inflammatory responses and expression of type 1 interferons (IFN1). In this report, we have explored the activity of the selective small molecule SUMOylation inhibitor subasumstat (TAK-981) in promoting antitumor innate immune responses. We demonstrate that treatment with TAK-981 results in IFN1-dependent macrophage and natural killer (NK) cell activation, promoting macrophage phagocytosis and NK cell cytotoxicity in ex vivo assays. Furthermore, pretreatment with TAK-981 enhanced macrophage phagocytosis or NK cell cytotoxicity against CD20+ target cells in combination with the anti-CD20 antibody rituximab. In vivo studies demonstrated enhanced antitumor activity of TAK-981 and rituximab in CD20+ lymphoma xenograft models. Combination of TAK-981 with anti-CD38 antibody daratumumab also resulted in enhanced antitumor activity. TAK-981 is currently being studied in phase 1 clinical trials (#NCT03648372, #NCT04074330, #NCT04776018, and #NCT04381650; www.clinicaltrials.gov) for the treatment of patients with lymphomas and solid tumors.
Introduction
Rituximab is an anti-CD20 therapeutic monoclonal immunoglobulin G1 (IgG1) antibody1 which is central to treatment of B-cell non-Hodgkin’s lymphoma (NHL). Multiple mechanisms of action have been reported for the antitumor activity of rituximab, including direct antiproliferative activity against, or induction of apoptosis in, CD20+ lymphoma cells, as well as Fc-mediated engagement of the innate immune system to promote antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), and complement-dependent cytotoxicity.2-7 Whereas the relative importance of the effector function of rituximab in clinical outcome has been debated, accumulating evidence supports a key role for ADCC and ADCP in mediating tumor cell killing.2,8 As a single agent or in combination with chemotherapy, rituximab is not curative in the majority of B-cell NHL patients, and the prognosis of relapsed/refractory patients is poor,9-12 highlighting the need for optimizing antitumor activity. Approaches for enhancing ADCC and ADCP have included glycoengineering or mutagenesis of the antibody fragment crystallizable (Fc) region to enhance interaction with Fc receptors expressed on innate immune cells13,14 or a combination of rituximab with agents that can stimulate innate immune cell effector function. Several reports have demonstrated that enhancement of macrophage phagocytosis by blocking the CD47-SIRPα axis can render tumor cells sensitive to monoclonal antibodies.15-19 In addition, stimulation of natural killer (NK) cell activity by lenalidomide and enhancement of ADCC in combination with rituximab has been implicated as one of the mechanistic rationales for clinical activity of the combination.20,21
Small ubiquitin-like modifier (SUMO) is a member of a ubiquitin-like protein superfamily. Subasumstat (TAK-981) is a potent and selective inhibitor of SUMOylation,22,23 a reversible posttranslational modification that regulates protein function by covalent attachment of SUMO protein to protein substrates.24 TAK-981 forms an irreversible adduct with each of the 3 functional mammalian SUMO paralogues (SUMO1, SUMO2, and SUMO3) when bound to the E1 SUMO-activating enzyme, preventing transfer of SUMO to substrates. As demonstrated by genetic inactivation of the pathway, inhibition of SUMOylation results in an inflammatory response dependent upon expression of type 1 interferons (IFN1) in mouse myeloid cells and human cell lines.25,26 The IFN1s are potent immunomodulatory molecules induced early in innate immune responses, which act on multiple cell types to shape both innate and adaptive antitumor immunity.27-30 In a recent study, we showed that pharmacological inhibition of SUMOylation with TAK-981 stimulated adaptive antitumor immune responses as a result of activation of IFN1 signaling in T cells and dendritic cells.23 In this study, we have examined the effect of TAK-981 on antitumor innate immune responses and documented IFN1-dependent phenotypic and functional activation of macrophages and NK cells ex vivo and in vivo. Moreover, combination of TAK-981 with rituximab was found to markedly enhance ADCP and ADCC, thereby augmenting antitumor activity of the therapeutic antibody rituximab and daratumumab.
Methods
Preparation of mouse and human macrophages, flow cytometry, macrophage phagocytosis assays, NK cytotoxicity assays, enzyme-linked immuno-sorbent assay, quantitative polymerase chain reaction (qPCR), RNA sequencing, and animal studies are described in supplemental Methods, available on the Blood Web site.
Compounds
TAK-981 was synthesized by Takeda Development Center Americas, Inc.22
Cell lines and culture
The following cell lines were used in this study: human Burkitt lymphoma Daudi (American Tissue Culture Collection [ATCC]), Daudi-KILR (DiscoveRx), human Burkitt lymphoma Raji (ATCC), human diffuse large B-cell lymphoma (DLBCL) OCI-Ly10 (University Health Network), human DLBCL TMD8 (Tokyo Medical and Dental University), human multiple myeloma LP-1 (Deutsche Sammlung von Mikroorganismen und Zellkulturen), and mouse B-cell lymphoma A20 (ATCC) cells. Cell lines were cultured at 37°C with 5% CO2 in the recommended medium supplemented with 10% to 20% fetal bovine serum (Sigma Aldrich). All cell lines were stocked after Mycoplasma testing and used within 2 months of resuscitation. No authentication was done by the authors.
Cell viability assay
Cell viability was assessed using the CellTiter-Glo luminescent cell viability assay (Promega) according to the manufacturer’s protocol.
Western blot analysis
Western blot analysis was performed as described in a previous study.31 The following antibodies were used for the western blot analysis: anti-STAT1 (#14994), -phospho-Tyr701-STAT1 (#9167), -ISG15 (#2743), -SUMO2/3 (#4971), and -GAPDH (#5174) (Cell Signaling Technology).
Statistical analysis
For comparison of 2 groups, significance was assessed by unpaired, 2-tailed Welch’s t tests (Prism v7.0 and v8.0, GraphPad). For in vivo experiments where treatment groups have different follow-up durations, the statistical analysis was performed at the time when all groups exist unless otherwise specified. To assess whether there is an additional benefit from combining 2 drug treatments, we tested the statistical significance of a synergy score, which is calculated by (where is the mean growth rate of the vehicle group, and are the mean growth rates of 2 single agent treatment groups, and is the mean growth rate of the corresponding combo treatment group). The P value of the synergy score was calculated by dividing the synergy score by its standard error and testing against a t distribution (2-tailed) with a corresponding degree of freedom estimated with the Welch-Satterthwaite equation. To assess the effect of treatment on survival curve, Weibull regression hazard ratio was determined unless otherwise specified. A P value < .05 was considered statistically significant.
Results
TAK-981 promotes IFN1-dependent macrophage M1 polarization and NK cell activation
Treatment of human monocyte–derived macrophages (hMDM) and mouse bone marrow–derived macrophages (mBMDM) with TAK-981 resulted in induction of IFNα/β and IP-10 as well as phosphorylation of STAT1 without a significant impact on cell viability (Figure 1A-B; supplemental Figure 1A-D), consistent with activation of IFN1 signaling. Macrophages have been broadly classified into 2 phenotypic states of polarization, proinflammatory M1 and anti-inflammatory M2, whose nomenclature is derived from the Th1 and Th2 cytokines that are associated with these macrophage phenotypes.32,33 To explore whether treatment with TAK-981 can modulate macrophage polarization, we examined the expression levels of M1 and M2 markers.32,33 Flow cytometry and qPCR analyses revealed that M1 markers (hMDM, CD80 and CD86; mBMDM, CD86 and inducible nitric oxide synthase [iNOS]) were induced by TAK-981 treatment (Figure 1C; supplemental Figure 1E-F). This was observed not only in the presence of M1-skewing factor IFNγ or lipopolysaccharide (LPS),32,33 but also in the presence of the M2-skewing factor interleukin-4 (IL-4) (Figure 1C; supplemental Figure 1E). Although CD86 is known as one of M1 markers, the induction of CD86 by IL-4 treatment was also observed in hMDM (Figure 1C), consistent with a previous observation,34 suggesting that CD86 alone does not serve as a definitive M1 marker in hMDM. iNOS expression in mBMDM was strongly induced only in the presence of LPS and was enhanced by TAK-981 treatment (supplemental Figure 1E), indicating that toll-like receptor 4–dependent induction of iNOS expression in mBMDM can be further enhanced by TAK-981. To further examine the effect of TAK-981 on macrophage polarization, we explored expression of high affinity immunoglobulin gamma Fc receptor I (FCGR1), shown to be highly expressed in human M1 macrophages.35,36 Flow cytometry analysis revealed that FCGR1 and another activating FCGR (hMDM, FCGR3; mBMDM, FCGR4) were induced by TAK-981 or IFNα2 treatment (Figure 1D; supplemental Figure 1G). Whereas treatment with TAK-981 upregulated mRNA levels of activating FCGRs, the upregulation in the mRNA levels of inhibitory FCGRs (hMDM, FCGR2B; mBMDM, Fcgr2) was not or minimally observed in qPCR and/or RNA sequencing analyses (supplemental Figure 1H-J). To assess the role of IFN1 signaling in macrophage responses to TAK-981, blockade of interferon alpha/beta receptor 1 (IFNAR1) or IFNAR2 was carried out using a neutralizing antibody. The increase in mRNA levels of CD86, FCGR1, and IP-10 in response to TAK-981 was suppressed by treatment with the anti-IFANR2 antibody in hMDM (Figure 1E), indicating dependency of induction on IFN1 signaling. However, induction of IFNβ expression by TAK-981 was not inhibited by treatment with anti-IFNAR2 antibody (Figure 1E), consistent with the observation that IFNβ is transcriptionally modulated by SUMOylation.25 The IFN1-dependent induction of M1 markers by TAK-981 was also confirmed at the protein level both in hMDM and mBMDM (Figure 1D; supplemental Figure 1G). Regarding M2 polarization in hMDM, treatment with TAK-981 suppressed IL-4–dependent induction of CD206 either when hMDM was treated with TAK-981 and IL-4 simultaneously or when hMDM was treated with IL-4 first followed by TAK-981 treatment (Figure 1F; supplemental Figure 1K), suggesting that TAK-981 can reverse phenotype of macrophages that have been already M2 polarized. In addition, treatment with TAK-981 suppressed induction of the anti-inflammatory chemokine CCL22 in hMDM (Figure 1G). Although IL-4–dependent induction of arginase, which is a well-known M2 marker in mBMDM, was not prevented by TAK-981 treatment in mBMDM, treatment with TAK-981 partially or completely suppressed upregulation of the expression of several other M2 markers including CD206 and CCL22 (supplemental Figure 1L), suggesting that an IFN1-independent signaling pathway may contribute to induction of some of the M2 markers. Collectively, our data demonstrate that TAK-981 enhances macrophage M1 polarization and at least partially suppresses M2 polarization through IFN1 pathway activation in both hMDM and mBMDM.
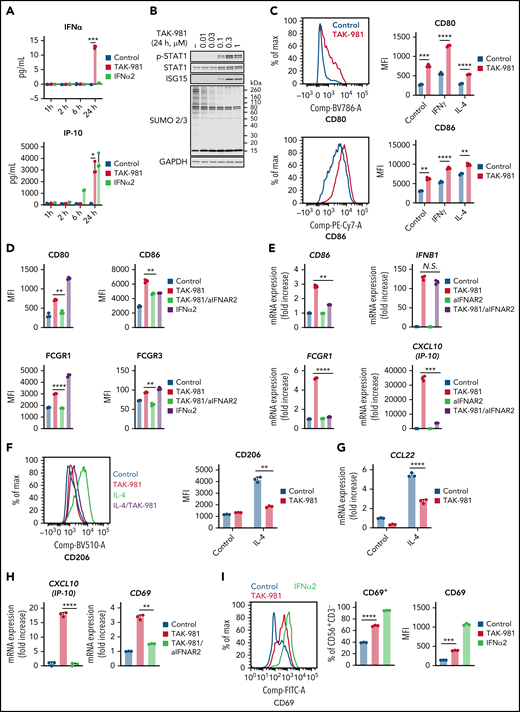
TAK-981 treatment enhances M1 polarization and NK activation in an IFN1-dependent manner. (A) Amount of IFNβ and IP-10 protein in the supernatant of 1 μM TAK-981- or 10 kU/mL IFNα2-treated hMDM at indicated time points (mean with SD; n = 3 biological replicates, 2-tailed unpaired Welch’s t test). (B) Western blots of indicated proteins in TAK-981–treated hMDM at 24 hours. (C) Representative flow cytometry histogram and median fluorescence intensity (MFI) of indicated proteins in 50 ng/mL IFNγ (M1 polarizing stimulation)-, 20 ng/mL IL-4 (M2 polarizing stimulation)-, and/or 1 μM TAK-981–treated hMDM at 48 hours (mean with SD; n = 3 biological replicates, 2-tailed unpaired Welch’s t test). (D) MFI of indicated proteins in 20 μg/mL anti-IFNAR2 antibody (aIFNAR2)-, 1 μM TAK-981-, and/or 10 kU/mL IFNα2-treated hMDM at 48 hours (mean with SD; n = 3 biological replicates, 2-tailed unpaired Welch’s t test). (E) mRNA expression of indicated genes in 20 μg/mL aIFNAR2- and/or 1 μM TAK-981–treated hMDM at 24 hours (mean with SD; n = 3 biological replicates, 2-tailed unpaired Welch’s t test). (F) Representative flow cytometry histogram and MFI of CD206 in 20 ng/mL IL-4- and/or 1 μM TAK-981–treated hMDM at 48 hours (mean with SD; n = 3 biological replicates, 2-tailed unpaired Welch’s t test). (G) mRNA expression of CCL22 in 20 ng/mL IL-4- and/or 1 μM TAK-981–treated hMDM at 24 hours (mean with SD; n = 3 biological replicates, 2-tailed unpaired Welch’s t test). (H) mRNA expression of IP-10 and CD69 in 20 μg/mL aIFNAR2- and/or 1 μM TAK-981–treated human NK cells at 24 hours (mean with SD; n = 3 biological replicates, 2-tailed unpaired Welch’s t test). (I) Representative flow cytometry histogram and percentage of CD69+ cells or MFI of CD69 in 1 μM TAK-981- or 10 kU/mL IFNα2-treated human NK cells at 24 hours (mean with SD; n = 3 biological replicates, 2-tailed unpaired Welch’s t test). *P < .05, **P < .01, ***P < .001, ****P < .0001. Experiment was repeated using hMDM or human NK cells generated from at least 2 healthy PBMC donors. mRNA, messenger RNA; N.S., not significant (P > .05); PBMC, peripheral blood mononuclear cells; SD, standard deviation.
TAK-981 treatment enhances M1 polarization and NK activation in an IFN1-dependent manner. (A) Amount of IFNβ and IP-10 protein in the supernatant of 1 μM TAK-981- or 10 kU/mL IFNα2-treated hMDM at indicated time points (mean with SD; n = 3 biological replicates, 2-tailed unpaired Welch’s t test). (B) Western blots of indicated proteins in TAK-981–treated hMDM at 24 hours. (C) Representative flow cytometry histogram and median fluorescence intensity (MFI) of indicated proteins in 50 ng/mL IFNγ (M1 polarizing stimulation)-, 20 ng/mL IL-4 (M2 polarizing stimulation)-, and/or 1 μM TAK-981–treated hMDM at 48 hours (mean with SD; n = 3 biological replicates, 2-tailed unpaired Welch’s t test). (D) MFI of indicated proteins in 20 μg/mL anti-IFNAR2 antibody (aIFNAR2)-, 1 μM TAK-981-, and/or 10 kU/mL IFNα2-treated hMDM at 48 hours (mean with SD; n = 3 biological replicates, 2-tailed unpaired Welch’s t test). (E) mRNA expression of indicated genes in 20 μg/mL aIFNAR2- and/or 1 μM TAK-981–treated hMDM at 24 hours (mean with SD; n = 3 biological replicates, 2-tailed unpaired Welch’s t test). (F) Representative flow cytometry histogram and MFI of CD206 in 20 ng/mL IL-4- and/or 1 μM TAK-981–treated hMDM at 48 hours (mean with SD; n = 3 biological replicates, 2-tailed unpaired Welch’s t test). (G) mRNA expression of CCL22 in 20 ng/mL IL-4- and/or 1 μM TAK-981–treated hMDM at 24 hours (mean with SD; n = 3 biological replicates, 2-tailed unpaired Welch’s t test). (H) mRNA expression of IP-10 and CD69 in 20 μg/mL aIFNAR2- and/or 1 μM TAK-981–treated human NK cells at 24 hours (mean with SD; n = 3 biological replicates, 2-tailed unpaired Welch’s t test). (I) Representative flow cytometry histogram and percentage of CD69+ cells or MFI of CD69 in 1 μM TAK-981- or 10 kU/mL IFNα2-treated human NK cells at 24 hours (mean with SD; n = 3 biological replicates, 2-tailed unpaired Welch’s t test). *P < .05, **P < .01, ***P < .001, ****P < .0001. Experiment was repeated using hMDM or human NK cells generated from at least 2 healthy PBMC donors. mRNA, messenger RNA; N.S., not significant (P > .05); PBMC, peripheral blood mononuclear cells; SD, standard deviation.
Like macrophages, NK cells are important effectors of innate immunity. TAK-981 treatment of NK cells isolated from human PBMCs resulted in a robust, and IFN1-dependent, increase in IP-10 expression without a significant impact on cell viability (Figure 1H; supplemental Figure 2A). Treatment of mouse splenic NK cells was also attempted, but ex vivo exposure to TAK-981 uniquely resulted in significant loss of viability in this cell population. In human NK cells, treatment with TAK-981 increased mRNA and protein levels of the early activation marker CD69 in an IFN1 signaling–dependent manner, and treatment with IFNα2 also induced CD69 (Figure 1H-I). Unlike macrophages, the expression levels of FCGR1 and FCGR3 were not increased by TAK-981 or IFNα2 treatment of human NK cells (supplemental Figure 2B-D).
TAK-981 enhances macrophage phagocytosis and NK cell cytotoxicity ex vivo in an IFN1-dependent manner
To explore whether TAK-981 modulated the activity of macrophages, phagocytosis assays were carried out using Daudi cells as target cells (treatment schematic shown in supplemental Figure 3A). In some experiments, Daudi cells expressing a luminescent protein (Daudi-KILR cells) were used to enable a high-throughput assay format and to validate the results in different readouts. Pretreatment of hMDM with TAK-981 or IFNα2 stimulated phagocytosis of Daudi cells (Figure 2A-B; supplemental Figure 3B). Because the compounds were washed out before coculture with the target cells, the enhancement of phagocytosis occurred due to direct effects on macrophages. Similarly, phagocytosis of Daudi-KILR cells was induced by TAK-981 treatment, irrespective of the presence of M1/M2 skewing factor IFNγ or IL-4 (Figure 2C). Because Daudi cells express CD20, the activity of the anti-CD20 antibody rituximab was tested in this assay to determine whether pretreatment of macrophages with TAK-981 can enhance ADCP. Pretreatment of macrophages with TAK-981 significantly enhanced the degree of target-cell phagocytosis achieved when rituximab was added to the coculture of macrophages and target cells, whereas the viability of nonphagocytosed target cells was not impacted (Figure 2A-C; supplemental Figure 3C). Notably, the enhancement of macrophage phagocytosis by TAK-981 was inhibited by combined treatment with anti-IFNAR2 antibody (Figure 2D). In contrast, direct treatment of the target cells with rituximab, TAK-981, or IFNα2 did not significantly affect cell viability (supplemental Figure 3D-E) and rituximab-mediated ADCP by macrophages (supplemental Figure 3F). As described for hMDM, treatment with TAK-981 also enhanced mBMDM phagocytosis in the presence of rituximab (supplemental Figure 3G). In order to determine whether TAK-981–mediated enhancement of ADCP could be achieved with a tumor-targeting antibody other than rituximab, we combined TAK-981 with daratumumab, an anti-CD38 antibody whose target is also expressed by Daudi cells.37 As shown in Figure 2E, treatment of hMDM with TAK-981 was also capable of enhancing daratumumab-mediated phagocytosis of Daudi-KILR cells in an IFNAR2-dependent manner. To determine whether TAK-981 similarly stimulates NK cell activity, NK cell cytotoxicity assays were carried out using Daudi cells or Daudi-KILR cells (see treatment schematic in supplemental Figure 3A). Treatment of human NK cells with TAK-981 or IFNα2 promoted NK cell–mediated killing of the target cells, which was further enhanced in the presence of rituximab, demonstrating stimulation of NK cell cytotoxicity and ADCC by TAK-981 (Figure 2F; supplemental Figure 3H-I). Moreover, IFNAR2 blockade suppressed the enhancement of NK cell cytotoxicity by TAK-981 (Figure 2F). Enhanced NK cell cytotoxicity/macrophage phagocytosis and ADCC/ADCP in response to TAK-981 treatment was also observed against Raji cells (supplemental Figure 3I-J).
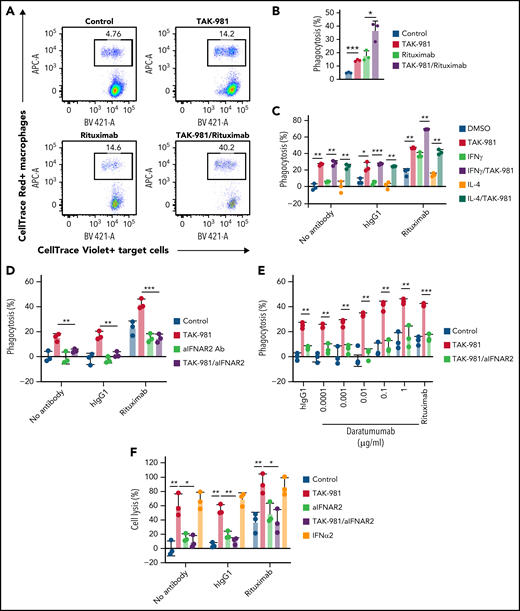
TAK-981 treatment augments rituximab-mediated macrophage phagocytosis and NK cell cytotoxicity in an IFN1-dependent manner. (A) Representative flow cytometry scatter plots. Macrophages are defined as CellTrace Red+ events and Daudi cells as CellTrace Violet+ events. Percent of CellTrace Red+Violet+ in CellTrace Violet+ events corresponds to percent of phagocytosed Daudi cells in total Daudi cells. (B) Phagocytic activity of 1 μM TAK-981–treated hMDM against Daudi cells in the presence of 1 μg/mL human IgG1 (hIgG1) or rituximab at an effector to target (E:T) ratio of 1:1 (mean with SD; n = 3 biological replicates, 2-tailed unpaired Welch’s t test). (C) Phagocytic activity of 50 ng/mL IFNγ-, 20 ng/mL IL-4-, and/or 1 μM TAK-981–treated hMDM against Daudi-KILR cells in the presence of hIgG1 or rituximab at an E:T ratio of 4:1 (mean with SD; n = 3 biological replicates, 2-tailed unpaired Welch’s t test). (D) Phagocytic activity of 20 μg/mL aIFNAR2- and/or 1 μM TAK-981–treated hMDM against Daudi-KILR cells in the presence of human IgG1 or rituximab at an E:T ratio of 4:1 (mean with SD; n = 3 biological replicates, 2-tailed unpaired Welch’s t test). (E) Phagocytic activity of 20 μg/mL aIFNAR2- and/or 1 μM TAK-981–treated hMDM against Daud-KILR cells in the presence of 1 μg/mL human IgG1, daratumumab, or 1 μg/mL rituximab at an E:T ratio of 4:1 (mean with SD; n = 3 biological replicates, 2-tailed unpaired Welch’s t test). (F) Cytotoxicity of 20 μg/ml aIFNAR2-, 1 μM TAK-981-, and/or 10 kU/mL IFNα2-treated human NK cells against Daudi-KILR cells in the presence of 1 μg/mL human IgG1 or 1 μg/mL rituximab at an E:T ratio of 4:1 (mean with SD; n = 3 biological replicates, 2-tailed unpaired Welch’s t test). *P < .05, **P < .01, ***P < .001. Experiment was repeated using hMDM or human NK cells generated from at least 2 healthy PBMC donors. SD, standard deviation; N.S., not significant (P > .05).
TAK-981 treatment augments rituximab-mediated macrophage phagocytosis and NK cell cytotoxicity in an IFN1-dependent manner. (A) Representative flow cytometry scatter plots. Macrophages are defined as CellTrace Red+ events and Daudi cells as CellTrace Violet+ events. Percent of CellTrace Red+Violet+ in CellTrace Violet+ events corresponds to percent of phagocytosed Daudi cells in total Daudi cells. (B) Phagocytic activity of 1 μM TAK-981–treated hMDM against Daudi cells in the presence of 1 μg/mL human IgG1 (hIgG1) or rituximab at an effector to target (E:T) ratio of 1:1 (mean with SD; n = 3 biological replicates, 2-tailed unpaired Welch’s t test). (C) Phagocytic activity of 50 ng/mL IFNγ-, 20 ng/mL IL-4-, and/or 1 μM TAK-981–treated hMDM against Daudi-KILR cells in the presence of hIgG1 or rituximab at an E:T ratio of 4:1 (mean with SD; n = 3 biological replicates, 2-tailed unpaired Welch’s t test). (D) Phagocytic activity of 20 μg/mL aIFNAR2- and/or 1 μM TAK-981–treated hMDM against Daudi-KILR cells in the presence of human IgG1 or rituximab at an E:T ratio of 4:1 (mean with SD; n = 3 biological replicates, 2-tailed unpaired Welch’s t test). (E) Phagocytic activity of 20 μg/mL aIFNAR2- and/or 1 μM TAK-981–treated hMDM against Daud-KILR cells in the presence of 1 μg/mL human IgG1, daratumumab, or 1 μg/mL rituximab at an E:T ratio of 4:1 (mean with SD; n = 3 biological replicates, 2-tailed unpaired Welch’s t test). (F) Cytotoxicity of 20 μg/ml aIFNAR2-, 1 μM TAK-981-, and/or 10 kU/mL IFNα2-treated human NK cells against Daudi-KILR cells in the presence of 1 μg/mL human IgG1 or 1 μg/mL rituximab at an E:T ratio of 4:1 (mean with SD; n = 3 biological replicates, 2-tailed unpaired Welch’s t test). *P < .05, **P < .01, ***P < .001. Experiment was repeated using hMDM or human NK cells generated from at least 2 healthy PBMC donors. SD, standard deviation; N.S., not significant (P > .05).
Antitumor activity of TAK-981 combined with rituximab or daratumumab in xenografts
The ability of TAK-981 to enhance ADCP and ADCC ex vivo encouraged us to explore whether TAK-981 could augment the in vivo antitumor activity of antibodies such as rituximab, which are dependent on these effector cells for their antitumor activity.2-5 As a starting point, we used the CD20- and CD38-expressing Burkitt lymphoma Daudi xenograft model grown in severe combined immunodeficiency (SCID) mice, which are deficient in T and B cells but retain myeloid cells and NK cells. Combination of TAK-981 with either rituximab or daratumumab resulted in synergistic combination activity in the Daudi model (Figure 3A). We also tested combination of TAK-981 with rituximab in 2 different CD20+ DLBCL xenograft models (OCI-Ly10 and TMD8). In the subcutaneous OCI-Ly10 xenograft models, whereas single-agent treatment with TAK-981 or rituximab markedly delayed tumor growth, combination treatment resulted in complete and durable elimination of palpable tumors (complete responses [CRs]) in all treated mice (Figure 3B). In the disseminated OCI-Ly10-luc xenograft models, single-agent treatment with TAK-981 did not have a significant impact on tumor growth and mouse survival, whereas the combined treatment with TAK-981 and rituximab demonstrated a more significant survival benefit than rituximab alone (supplemental Figure 4A-C). Another clinically approved anti-CD20 antibody, obinutuzumab, showed robust antitumor activity in combination with TAK-981, which is comparable to that of TAK-981 and rituximab combination (Figure 3C). In the TMD8 xenograft model, TAK-981 had minimal effect on tumor growth, and rituximab treatment resulted in tumor growth delay (Figure 3D). However, the combined treatment of TAK-981 and rituximab induced tumor regression and extended survival of mice (Figure 3D; supplemental Figure 5A). In addition to cell line xenograft models, we also tested a human primary CD20+ DLBCL xenograft model, PHTX-166L. The tumors showed comparable tumor growth inhibition in response to treatment with either single agent, whereas the combination resulted in synergistic tumor growth inhibition including 1 CR out of 6 mice (Figure 3E). The LP-1 multiple myeloma xenograft model was also investigated in SCID mice to explore TAK-981 combination activity with daratumumab in a myeloma setting. The combination of TAK-981 with daratumumab resulted in tumor stasis and prolonged survival, in contrast to only minimal activity in response to either single agent (Figure 3F; supplemental Figure 5B). To examine the in vivo combination activity in an immune-competent setting, we generated a mouse syngeneic model bearing mouse A20 B-cell lymphoma tumor cells engineered to express human CD38. Whereas TAK-981 or daratumumab had minimal effect on survival, combined treatment of TAK-981 and daratumumab significantly extended survival (supplemental Figure 6A-B). Marked body weight loss was not induced by the treatments throughout the study period (supplemental Figures 6C and 7A-G).
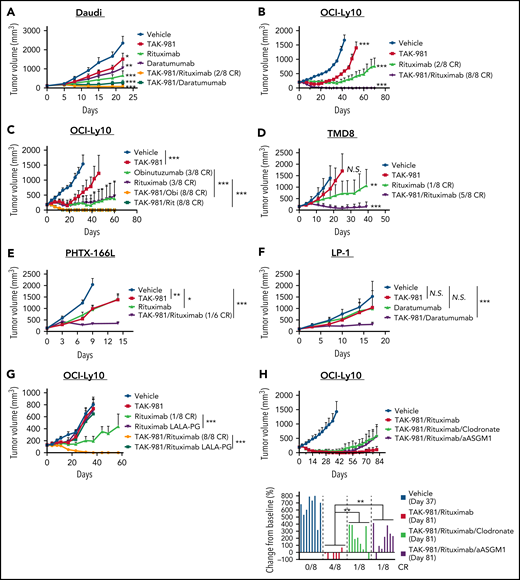
Antitumor activity of TAK-981 combined with monoclonal antibodies in xenograft models. (A) Tumor volumes in Daudi-bearing mice treated with TAK-981 (7.5 mg/kg, biweekly), rituximab (3 mg/kg, weekly), and/or daratumumab (7.5 mg/kg, biweekly) as indicated for 3 weeks (mean with SD; n = 8 mice per group in the representative study, 2-tailed unpaired Welch’s t test). The P value of the synergy score was P < .001. (B) Tumor volumes in OCI-Ly10–bearing mice treated with TAK-981 (7.5 mg/kg, biweekly) and/or rituximab (1 mg/kg, weekly) as indicated for 2 weeks (mean with SD; n = 8 mice per group in the representative study, 2-tailed unpaired Welch’s t test). The P value of the synergy score was P < .001. (C) Tumor volumes in OCI-Ly10–bearing mice treated with TAK-981 (7.5 mg/kg, biweekly), obinutuzumab (Obi, 1 mg/kg, weekly), and/or rituximab (Rit, 1 mg/kg, weekly) as indicated for 2 weeks (mean with SD; n = 8 mice per group in a single experiment, 2-tailed unpaired Welch’s t test). (D) Tumor volumes in TMD8-bearing mice treated with TAK-981 (15 mg/kg, biweekly) and/or rituximab (3 mg/kg, weekly) as indicated for 2 weeks (mean with SD; n = 8 mice per group in the representative study, 2-tailed unpaired Welch’s t test). The P value of the synergy score was P = .08 (additive combination effect). (E) Tumor volumes in PHTX-166L–bearing mice treated with TAK-981 (5 mg/kg, weekly) and/or rituximab (5 mg/kg, weekly) as indicated for 2 weeks (mean with SD; n = 6 mice per group in the representative study, 2-tailed unpaired Welch’s t test). The P value of the synergy score was P < .05. (F) Tumor volumes in LP-1–bearing mice treated with TAK-981 (7.5 mg/kg, biweekly) and/or daratumumab (2.5 mg/kg, biweekly) as indicated for 5 weeks (mean with SD; n = 8 mice per group in the representative study, 2-tailed unpaired Welch’s t test). The P value of the synergy score was P < .05. (G) Tumor volumes in OCI-Ly10–bearing mice treated with TAK-981 (7.5 mg/kg, biweekly), rituximab (3 mg/kg, weekly), and/or rituximab with LALA-PG mutations (3 mg/kg, weekly) as indicated for 2 weeks (mean with SD; n = 8 mice per group in a single experiment, 2-tailed unpaired Welch’s t test). (H) Tumor volumes in OCI-Ly10–bearing mice were treated with TAK-981 (7.5 mg/kg, biweekly), rituximab (1 mg/kg, weekly), clodronate liposome (200 µL per mouse at a first dose and 100 µL per mouse at subsequent doses, biweekly), and/or anti-asialo GM1 (aASGM1, 0.25 mg per mouse, weekly) as indicated for 2 weeks (mean with SD; n = 8 mice per group in the representative study). Bar graphs show percent change in tumor volumes from baseline on the indicated day of measurement (individual value; n = 8 mice per group in the representative study, 2-tailed unpaired Welch’s t test). *P < .05, **P < .01, ***P < .001. The number of mice achieved CR per the number of total mice in the group is shown. At least 2 similar experiments were performed unless otherwise specified. SD, standard deviation; N.S., not significant (P > .05).
Antitumor activity of TAK-981 combined with monoclonal antibodies in xenograft models. (A) Tumor volumes in Daudi-bearing mice treated with TAK-981 (7.5 mg/kg, biweekly), rituximab (3 mg/kg, weekly), and/or daratumumab (7.5 mg/kg, biweekly) as indicated for 3 weeks (mean with SD; n = 8 mice per group in the representative study, 2-tailed unpaired Welch’s t test). The P value of the synergy score was P < .001. (B) Tumor volumes in OCI-Ly10–bearing mice treated with TAK-981 (7.5 mg/kg, biweekly) and/or rituximab (1 mg/kg, weekly) as indicated for 2 weeks (mean with SD; n = 8 mice per group in the representative study, 2-tailed unpaired Welch’s t test). The P value of the synergy score was P < .001. (C) Tumor volumes in OCI-Ly10–bearing mice treated with TAK-981 (7.5 mg/kg, biweekly), obinutuzumab (Obi, 1 mg/kg, weekly), and/or rituximab (Rit, 1 mg/kg, weekly) as indicated for 2 weeks (mean with SD; n = 8 mice per group in a single experiment, 2-tailed unpaired Welch’s t test). (D) Tumor volumes in TMD8-bearing mice treated with TAK-981 (15 mg/kg, biweekly) and/or rituximab (3 mg/kg, weekly) as indicated for 2 weeks (mean with SD; n = 8 mice per group in the representative study, 2-tailed unpaired Welch’s t test). The P value of the synergy score was P = .08 (additive combination effect). (E) Tumor volumes in PHTX-166L–bearing mice treated with TAK-981 (5 mg/kg, weekly) and/or rituximab (5 mg/kg, weekly) as indicated for 2 weeks (mean with SD; n = 6 mice per group in the representative study, 2-tailed unpaired Welch’s t test). The P value of the synergy score was P < .05. (F) Tumor volumes in LP-1–bearing mice treated with TAK-981 (7.5 mg/kg, biweekly) and/or daratumumab (2.5 mg/kg, biweekly) as indicated for 5 weeks (mean with SD; n = 8 mice per group in the representative study, 2-tailed unpaired Welch’s t test). The P value of the synergy score was P < .05. (G) Tumor volumes in OCI-Ly10–bearing mice treated with TAK-981 (7.5 mg/kg, biweekly), rituximab (3 mg/kg, weekly), and/or rituximab with LALA-PG mutations (3 mg/kg, weekly) as indicated for 2 weeks (mean with SD; n = 8 mice per group in a single experiment, 2-tailed unpaired Welch’s t test). (H) Tumor volumes in OCI-Ly10–bearing mice were treated with TAK-981 (7.5 mg/kg, biweekly), rituximab (1 mg/kg, weekly), clodronate liposome (200 µL per mouse at a first dose and 100 µL per mouse at subsequent doses, biweekly), and/or anti-asialo GM1 (aASGM1, 0.25 mg per mouse, weekly) as indicated for 2 weeks (mean with SD; n = 8 mice per group in the representative study). Bar graphs show percent change in tumor volumes from baseline on the indicated day of measurement (individual value; n = 8 mice per group in the representative study, 2-tailed unpaired Welch’s t test). *P < .05, **P < .01, ***P < .001. The number of mice achieved CR per the number of total mice in the group is shown. At least 2 similar experiments were performed unless otherwise specified. SD, standard deviation; N.S., not significant (P > .05).
The in vitro viability of Daudi, OCI-Ly10, and TMD8 did not differ in response to either single-agent or combination treatment with TAK-981 and rituximab, suggesting that a direct effect of TAK-981/rituximab combination on tumor cells is not driving the increased in vivo antitumor efficacy observed (supplemental Figure 8). To assess the contribution of effector function to rituximab-mediated antitumor activity, we generated an effector-attenuated rituximab mutant with L234A, L235A, and P329G (LALA-PG) mutations engineered into the Fc region of the antibody.38,39 We validated the binding activity of rituximab LALA-PG to CD20 (expressed on Daudi cells) and the loss of effector activity of rituximab LALA-PG in vitro in an ADCC assay (supplemental Figure 9A-B). In the OCI-Ly10 xenograft model, LALA-PG–mutated rituximab did not demonstrate antitumor activity either as a single agent or in combination with TAK-981 (Figure 3G), indicating that rituximab effector function is critical for antitumor activity and combination with TAK-981. To better understand the mechanism of the in vivo antitumor activity of TAK-981 combined with rituximab, we assessed the effect of depleting macrophages with clodronate or NK cells with anti-asialo GM1.3,39 Anti-asialo GM1-mediated NK cell depletion in blood and clodronate-mediated macrophage depletion in tumors were confirmed by flow cytometry (supplemental Figure 10A-B). Treatment with clodronate or anti-asialo GM1 partially abrogated the antitumor activity of combined treatment with TAK-981 and rituximab in OCI-Ly10 xenografts (Figure 3H). Notably, combined treatment with clodronate and anti-asialo GM1 more rapidly abrogated the antitumor activity of TAK-981/rituximab combination (supplemental Figure 11A). Flow cytometric analysis confirmed the in vivo codepletion of macrophages and NK cells by the combined treatment with clodronate and anti-asialo GM1, though NK cell depletion by anti-asialo GM1 treatment was somewhat less effective when combined with clodronate treatment (supplemental Figure 11B-C), perhaps reflecting compromised ADCP of NK cells as a result of macrophage reduction. In the TMD8 xenograft model, the antitumor activity of TAK-981/rituximab combination was also reversed by combined treatment with clodronate and anti-asialo GM1 (supplemental Figure 11D). Furthermore, rituximab did not show antitumor activity either as a single agent or in combination with TAK-981 in severely immune-deficient mice bearing OCI-Ly10 xenografts, in which macrophages and NK cells are defective (nonobese diabetic/SCID) or macrophages are defective and NK cells are absent (nonobese diabetic/SCID/Interleukin-2Rγnull) (supplemental Figure 12). Although complement-dependent cytotoxicity has been implicated in mediating rituximab antitumor activity in some settings, complement depletion by cobra venom factor did not abrogate the antitumor activity of combined treatment with TAK-981 and rituximab (supplemental Figure 13A-B). Collectively, these results indicate a key role for macrophages and NK cells in driving the antitumor activity of combination treatment with TAK-981 and rituximab.
TAK-981 promotes macrophage M1 polarization and NK cell activation in CD20+ lymphoma xenografts
Tumor immunophenotyping analysis was performed to explore the impact of TAK-981 treatment on the tumor microenvironment in the OCI-Ly10 xenograft model (Figure 4A; supplemental Figure 14). Flow cytometry analysis revealed a significant decrease in the proportion of CD11b+Ly6G-Ly6ClowF4/80+ tumor-associated macrophages (TAM) in response to TAK-981 treatment on both days 5 and 9 after initiation of treatment (Figure 4B-C). The proportion of NK cells in tumors was seen to increase on both days in TAK-981–treated mice (Figure 4B-C). In contrast, the proportion of CD11b+Ly6G-Ly6Chigh monocytes or CD11b+Ly6G+Ly6Cint neutrophils was increased or decreased by TAK-981 treatment only on day 9, respectively (Figure 4B-C). Given the earlier and greater changes observed on day 5 post–TAK-981 treatment, the changes in macrophages and NK cells would be primary events. These changes were not the results of just redistribution between proportions, because the absolute number of the cells adjusted to tumor weight showed comparable tendency (supplemental Figure 15). Notably, although the proportion of M2-like TAM (CD86−/CD206+) were decreased by TAK-981 and further decreased by the combination treatment with rituximab, that of M1-like TAM (CD86+/CD206−) were increased by the treatments (Figure 4D). Moreover, treatment with TAK-981 increased the proportion of FCGR4+ TAM as well as that of IFNγ+ NK cells, with a trend toward increased CD69+ NK cells in the TAK-981− and/or combination-treated tumors (Figure 4E-F).
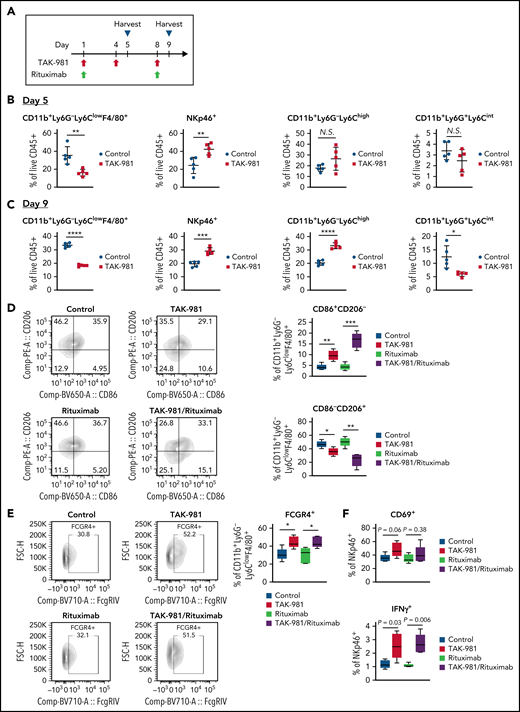
TAK-981 treatment increases M1-like/M2-like ratio in TAM and activates tumor-infiltrated NK cells in OCI-Ly10 xenografts. (A) Schematic diagram of study design. Tumors from 7.5 mg/kg TAK-981 (administered on day 1, 4, and 8)- and/or 1 mg/kg rituximab (administered on day 1 and 8)- treated OCI-Ly10 tumor-bearing mice on day 5 or day 9 were analyzed. (B) Proportion of CD11b+Ly6G−Ly6ClowF4/80+ TAM, NKp46+ NK cells, CD11b+Ly6G−Ly6Chigh monocytes, or CD11b+Ly6G+Ly6Cint neutrophils on day 5 (mean with SD; n = 5 mice per group in the representative study, 2-tailed unpaired Welch’s t test). (C) Proportion of CD11b+Ly6G−Ly6ClowF4/80+ TAM, NKp46+ NK cells, CD11b+Ly6G−Ly6Chigh monocytes, or CD11b+Ly6G+Ly6Cint neutrophils on day 9 (mean with SD; n = 5 mice per group, 2-tailed unpaired Welch’s t test). (D) Left, representative flow cytometry scatter plots of CD86/CD206. Right, proportion of M1-like (CD86+CD206−) TAM or M2-like (CD86−CD206+) TAM in tumors on day 5. (E) Left, representative flow cytometry scatter plots of FCGR4/FSC-H. Right, proportion of FCGR4+ TAM in tumors on day 5. (F) Proportion of CD69+ or IFNγ+ NK cells in tumors on day 5. Box plot center, box, and whiskers correspond to median, IQR (interquartile range), and 1.5 times IQR, respectively (n = 5 mice per group in the representative study, 2-tailed unpaired Welch’s t test). Two similar experiments were performed. SD, standard deviation.
TAK-981 treatment increases M1-like/M2-like ratio in TAM and activates tumor-infiltrated NK cells in OCI-Ly10 xenografts. (A) Schematic diagram of study design. Tumors from 7.5 mg/kg TAK-981 (administered on day 1, 4, and 8)- and/or 1 mg/kg rituximab (administered on day 1 and 8)- treated OCI-Ly10 tumor-bearing mice on day 5 or day 9 were analyzed. (B) Proportion of CD11b+Ly6G−Ly6ClowF4/80+ TAM, NKp46+ NK cells, CD11b+Ly6G−Ly6Chigh monocytes, or CD11b+Ly6G+Ly6Cint neutrophils on day 5 (mean with SD; n = 5 mice per group in the representative study, 2-tailed unpaired Welch’s t test). (C) Proportion of CD11b+Ly6G−Ly6ClowF4/80+ TAM, NKp46+ NK cells, CD11b+Ly6G−Ly6Chigh monocytes, or CD11b+Ly6G+Ly6Cint neutrophils on day 9 (mean with SD; n = 5 mice per group, 2-tailed unpaired Welch’s t test). (D) Left, representative flow cytometry scatter plots of CD86/CD206. Right, proportion of M1-like (CD86+CD206−) TAM or M2-like (CD86−CD206+) TAM in tumors on day 5. (E) Left, representative flow cytometry scatter plots of FCGR4/FSC-H. Right, proportion of FCGR4+ TAM in tumors on day 5. (F) Proportion of CD69+ or IFNγ+ NK cells in tumors on day 5. Box plot center, box, and whiskers correspond to median, IQR (interquartile range), and 1.5 times IQR, respectively (n = 5 mice per group in the representative study, 2-tailed unpaired Welch’s t test). Two similar experiments were performed. SD, standard deviation.
Discussion
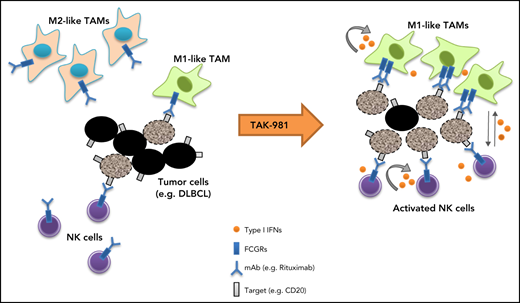
Our studies indicate that the SUMOylation inhibitor TAK-981 stimulates macrophage and NK cell function through IFN1 pathway activation, leading to enhanced antitumor activity in combination with tumor-targeting monoclonal antibodies such as rituximab and daratumumab (Figure 5). Both ex vivo and in vivo studies support a central dependency of the observed combination activity on the enhanced function of macrophages and NK cells as a result of IFN1 pathway activation. These observations extend previous work documenting IFN1-dependent activation of dendritic cells and T cells by TAK-981,23 indicative of broad IFN1-dependent immune cell activation by TAK-981.
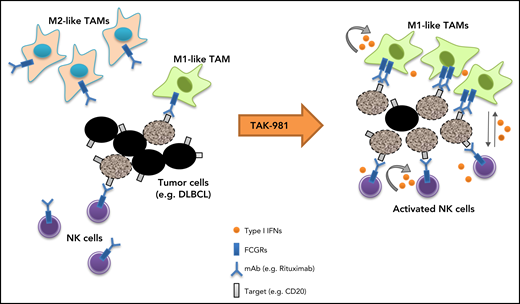
Schematic model of TAK-981–mediated antitumor innate immune responses through IFN1 activation. TAK-981 treatment enhances M1 macrophage polarization and NK cell activation through IFN1 signaling, leading to enhanced macrophage phagocytosis and NK cell cytotoxicity against tumor cells, particularly in the presence of therapeutic monoclonal antibodies like rituximab. The induction of activating FCGR levels in macrophages following TAK-981 treatment may also contribute to the enhanced macrophage phagocytosis.
Schematic model of TAK-981–mediated antitumor innate immune responses through IFN1 activation. TAK-981 treatment enhances M1 macrophage polarization and NK cell activation through IFN1 signaling, leading to enhanced macrophage phagocytosis and NK cell cytotoxicity against tumor cells, particularly in the presence of therapeutic monoclonal antibodies like rituximab. The induction of activating FCGR levels in macrophages following TAK-981 treatment may also contribute to the enhanced macrophage phagocytosis.
IFN1 acts directly on NK cells to promote their activation, proliferation, and cytotoxic function.40 Recently, induction of IFNβ by agonism of the cyclic GMP-AMP synthase-stimulator of interferon genes protein pathway has been demonstrated to trigger NK cell–mediated tumor killing.27 In contrast to NK cells, the role of IFN1 in macrophage polarization phenotypes has been less clearly defined. It is important to note that although the status of TAMs is often characterized by a binary M1-M2 polarization designation, it has been demonstrated that tumor macrophage activation states represent a continuum of phenotypes rather than 2 binary states. Nevertheless, although a simplification, the M1/M2 designation provides a shorthand for distinguishing between classically (proinflammatory and antitumorigenic) and alternatively (anti-inflammatory and protumorigenic) activated macrophages, respectively.41,42 In certain microbial infections such as tuberculosis, treatment with IFN1 has an anti-inflammatory effect on macrophages both in mouse and human.43 On the other hand, it has been reported that IFN1 signaling mediates LPS-induced expression of the M1 marker iNOS and synergizes with toll-like receptor agonists to activate M1 macrophages in mBMDM.44,45 Whereas IFN1 is known to be highly expressed in M1 macrophages and contributes to M1 polarization in human cell line–derived macrophages,46,47 the roles of IFN1 in human primary macrophage polarization have been less well characterized. Our data indicates that IFN1 pathway activation by TAK-981 induces proinflammatory M1 marker expression both in mBMDM and hMDM, and that this can be markedly enhanced by coadministration of LPS and/or IFNγ. It is possible that the context-dependent effect of IFN1 on macrophage activity reflects the impact of environmental cues on remodeling the macrophage enhancer repertoire. Interestingly, SUMO is an integral component of chromatin and, as previously mentioned, shown to play a role in modulating IFN1 expression via inhibitory SUMOylation of a distal IFNβ enhancer element.25 Although our ex vivo data indicates that TAK-981 treatment enhances macrophage M1 polarization in an IFNAR-dependent manner, it is also possible that global suppression of SUMOylation in enhancer regions of genes encoding proinflammatory proteins, and/or in transcription factors mobilized by IFN1, facilitates and contributes to IFN1-dependent responses induced by TAK-981.
The in vivo mechanisms of tumor cell depletion documented for rituximab are variable and likely, at least in part, dependent on the tumor model used. In our study, we have almost exclusively used human xenograft tumors grown in SCID mice, lacking adaptive immunity, so as to focus on the effects of TAK-981 on the contribution of innate immune responses to treatment with rituximab. Antitumor activity of both single-agent and combination treatment with rituximab was abrogated upon use of the effector-attenuated rituximab LALA-PG mutant, indicating that Fc-mediated engagement of effector cells is critical. Notably, the marked enhancement of antitumor activity achieved by combined treatment with TAK-981 and rituximab was partially reversed by macrophage or NK cell depletion, indicative of a key role for these effector cells in the antitumor combination activity. The incomplete rescue of tumor growth by macrophage or NK cell depletion may reflect a need for depletion of both cell types, or a preferential dependency on macrophage depletion, which is incomplete in tumors following clodronate treatment, as previously documented.48-50 Macrophage depletion by other approaches such as CSF1R inhibitors may help further clarify the importance of macrophages in future studies.
TAK-981 treatment not only induced proinflammatory activation of macrophages but also increased FCGR expression in an IFN1-dependent manner in macrophages, which may help enhance ADCP. This is consistent with published reports of STING agonism eliciting increased FCGR expression through IFN1 production, in both hMDM and mBMDM, thereby augmenting ADCP.51 Induction of FCGR expression was not observed in NK cells following TAK-981 treatment, indicating that TAK-981 may stimulate cell cytotoxicity/ADCC of NK cells independent of FCGR upregulation. A caveat of the ex vivo assay is that it is challenging to distinguish between enhancement of ADCC/ADCP by TAK-981 vs additive readout of TAK-981–induced NK cell cytotoxicity/macrophage phagocytosis in combination with ADCC/ADCP. Nevertheless, our in vivo study demonstrates clear enhancement of rituximab and daratumumab antitumor activity when combined with TAK-981 in multiple preclinical models, indicating the robust potential of the combination for the treatment of cancer.
We found that TAK-981 induces M1 polarization and enhances macrophage phagocytosis/ADCP. It has been controversial whether the polarization state of macrophages influences ADCP response.48,52,53 In our study, M1 macrophages showed higher levels of phagocytosis/ADCP than M2 macrophages. More importantly, TAK-981 was able to enhance phagocytosis/ADCP regardless of polarization conditions in the ex vivo assay. In vivo, there were more M1-like TAM and less M2-like TAM in the tumors post–TAK-981 treatment. Although the proportion and number of macrophages were decreased up to threefold, we hypothesize there was nevertheless a qualitative shift toward M1 polarization that would be predicted to have tumoricidal activity and generate a more proinflammatory tumor microenvironment, favorable to activation of NK cells. In addition, the upregulation of activating FCGR by TAK-981 may contribute to the enhancement of ADCP. Tumor immunophenotyping analysis also revealed that other innate immune cell subsets such as neutrophils and monocytes were affected by TAK-981 treatment in the OCI-Ly10 tumors, though the changes are likely to be secondary events. Further investigation should allow us to better understand the impact of TAK-981 on these immune cells in tumor microenvironment.
Our previous data has shown that TAK-981 induces transient B-cell depletion in mice,23 raising the possibility that intrinsic sensitivity of B lineage cells to TAK-981 may contribute to antitumor activity in the preclinical models. Indeed, we have observed moderate and variable single-agent in vitro and in vivo activity of TAK-981 in the B-cell lymphoma cell lines. It is possible that a direct effect of TAK-981 on cancer cells contributes to the enhanced antitumor activity when combined with rituximab or daratumumab. Generation of cell lines that are insensitive to TAK-981 could be informative in this regard.
We have shown here that TAK-981 can enhance the activity of both macrophages and NK cells and demonstrated that IFN1-dependent stimulation of macrophage and NK cell activity is a key mechanism of action underlying antitumor combination activity with rituximab or daratumumab. Our findings establish a preclinical rationale for targeting SUMOylation using small-molecule inhibitors to activate IFN1 signaling in combination with tumor-targeting antibodies such as rituximab and daratumumab for the treatment of cancer. TAK-981 is being investigated in combination with rituximab in patients with relapsed/refractory CD20+ NHL (NCT04074330; clinicaltrials.gov) and in combination with anti-CD38 monoclonal antibodies in a phase 1/2 study in patients with multiple myeloma (NCT04776018; clinicaltrials.gov), as well as a single agent in a phase 1/2 clinical study in patients with advanced or metastatic solid tumors or relapsed/refractory hematologic malignancies (NCT03648372; clinicaltrials.gov), and in combination with pembrolizumab in a phase 1/2 study in patients with advanced or metastatic solid tumors (NCT04381650; clinicaltrials.gov).
Acknowledgments
The authors acknowledge University Health Network for the OCI-Ly10 cell line; Tokyo Medical and Dental University for the TMD8 cell line; Denise Driscoll for the advice on ADCP assay; Cierra Casson and Gurpanna Saggu for the advice and help on macrophage experiments; Cong Li, Min Young Lee, and Eric Lightcap for the advice on data and statistical analyses; and Katherine Galvin and Igor Proscurshim for the manuscript review.
Authorship
Contribution: A.N., A.B., G.S., and D.H. designed research; A.N., S.G., K.S., K.X., Y.Z., and D.C. performed research; A.N., S.G., K.S., K.X., and D.C. analyzed data; and A.N. and D.H. wrote the paper.
Conflict-of-interest disclosure: All authors were employees of Millennium Pharmaceuticals Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited, during the course of this study. The legal name has recently changed to Takeda Development Center Americas, Inc.
The current affiliation for G.S. is Affini-T Therapeutics, Inc., Needham, MA.
The current affiliation for D.H. is Jounce Therapeutics, Cambridge, MA.
Correspondence: Akito Nakamura, Oncology Drug Discovery Unit, Takeda Development Center Americas, Inc., 40 Landsdowne St, Cambridge, MA 02139; e-mail: akt.nakamura@gmail.com; and Allison Berger, Oncology Therapeutic Area Unit, Takeda Development Center Americas, Inc., 40 Landsdowne St, Cambridge, MA 02139; e-mail: allison.berger@takeda.com.
RNA sequencing data are deposited in the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) under accession number GSE185841. All data associated with this study are present in the paper or supplemental Materials. TAK-981 is available to academic researchers upon completion of a materials transfer agreement with Takeda Development Center Americas, Inc.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.