

In this issue of Blood, Li et al provide experimental evidence that the high mobility group A1 (HMGA1) gene plays an important role in progression of Philadelphia-negative myeloproliferative neoplasms (MPNs) toward acute myeloid leukemia (AML).1
The HMGA gene family is composed of HMGA1 (or HMG-I/Y) located on chromosome 6p21 and HMGA2 (or HMGI-C) located on 12q13-15 encoding for non-histone AT hook-containing nuclear proteins that control chromatin architecture and gene expression by binding to the minor groove of AT-rich regions in double-stranded DNA. Physiologically, HMGA proteins are abundant and critical for normal embryonic development, whereas they are almost absent in adult tissues. Multiple studies have shown aberrant expression of HMGA family proteins in a wide variety of experimental and primary human cancers, including hematologic malignancies.2 Previous studies suggested that aberrantly high expression of HMGA2 contributes to increased proliferation of JAK2V617F+ MPN cells. Elevated HMGA2 expression in MPNs was shown to be the consequence of chromosomal lesions that lead to loss of the 3′ untranslated region targeted by the regulatory let-7 microRNA (mRNA) or of mutations in the epigenetic regulators EZH2 or ASXL disrupting Polycomb-mediated gene repression.3
Li et al found increased HMGA1 mRNA expression in primary CD34+ cells from patients with JAK2V617F+ MPNs that progressed to AML (see figure). Experimental HMGA1 gene silencing impaired proliferation, colony formation, and in vivo disease propagation by 3 secondary AML cell lines. In addition, Hmga1+/− heterozygous JAK2V617F+ mice showed a significantly delayed progression of MPNs to myelofibrosis (MF) compared with controls. Moreover, mice transplanted with JAK2V617F-expressing Hmga1+/− bone marrow (BM) developed a much milder disease than controls. Integrative epigenomic analysis of AML cell lines upon HMGA1 silencing revealed a significant negative correlation with the expression of target genes of the GATA2 stemness regulator, and HMGA1 knockdown reduced GATA2 mRNA and protein expression. HMGA1 seems to control GATA2 expression by binding to a previously identified +9.5-kB enhancer element. Notably, GATA2 knockdown phenocopied the effects of reduced HMGA1 expression in vitro and in vivo, whereas GATA2 overexpression partially rescued the antileukemic effects of HMGA1 silencing in DAMI and SET-2 AML cells. RNA sequencing of matched patients with MF that progressed to AML revealed increased expression of HMGA1 and GATA2 associated with increased expression of genes that regulate cell cycle progression, DNA repair, and metabolism. Collectively, Li et al unraveled a novel HMGA1-controlled transcriptional network involving GATA2 that is active in JAK2V617F+ MPNs and important for maintaining the transformed phenotype of secondary AML cell lines.
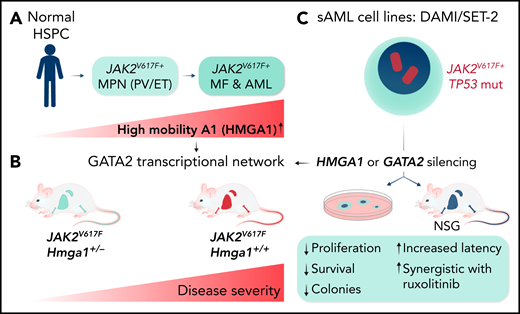
Experiments revealing a role of HMGA1 in MPN progression. (A) Increased expression of HMGA1 in primary hematopoietic cells from patients with MPN progressing to MF and AML. (B) Hmga1+/− mice developed a significantly milder disease phenotype than Hmga1+/+JAK2V617F transgenic mice. (C) Gene silencing studies revealed the existence of a GATA2-controlled transcriptional network involved in maintenance of a transformed phenotype of secondary JAK2V617F+ AML cell lines (SET-2, DAMI). ET, essential thrombocytopenia; HSPC, hematopoietic stem and progenitor cell; mut, mutated; PV, polycythemia vera; sAML, secondary AML. Professional illustration by Somersault18:24.
Experiments revealing a role of HMGA1 in MPN progression. (A) Increased expression of HMGA1 in primary hematopoietic cells from patients with MPN progressing to MF and AML. (B) Hmga1+/− mice developed a significantly milder disease phenotype than Hmga1+/+JAK2V617F transgenic mice. (C) Gene silencing studies revealed the existence of a GATA2-controlled transcriptional network involved in maintenance of a transformed phenotype of secondary JAK2V617F+ AML cell lines (SET-2, DAMI). ET, essential thrombocytopenia; HSPC, hematopoietic stem and progenitor cell; mut, mutated; PV, polycythemia vera; sAML, secondary AML. Professional illustration by Somersault18:24.
Li et al found that HMGA1 silencing did not alter low-level HMGA2 expression in secondary AML cell lines. In addition, the authors previously showed that heterozygous or homozygous loss of Hmga2 did not affect the phenotype of JAK2V617F transgenic mice.4 Notably, Li et al characterized HMGA1 by using a loss-of-function approach, whereas previous studies characterized HMGA2 as a functional collaborator of JAK2V617F in mostly gain-of-function experiments.
Although Li et al show that HMGA1 contributes to the maintenance of a transformed phenotype of secondary AML cell lines and to the disease severity in JAK2V617F transgenic mice, the detailed contribution of HMGA1 in MPN progression remains unclear. Because progression toward AML may include multiple genetic events, modeling it is challenging. Nevertheless, previous studies have shown that experimental loss of TP53 is sufficient to induce blast-phase transformation of JAK2V617F+ MPNs.5 Because the AML cells lines used by Li et al all carry JAK2V617F and TP53 mutations, one wonders about the role of the latter on increased HMGA1 expression. Earlier work in solid cancer cells suggested that there are multiple potential functional interactions, including TP53-controlled expression of mRNAs targeting HMGA genes.6 Future studies will need to investigate whether, similar to what has been observed in solid cancers, HMGA1 may exert its leukemogenic effect not only as a nuclear chromatin architecture protein but also as secreted factor binding to advanced glycosylation end product-specific receptor–inducing diverse intracellular signaling pathways.7
Li et al observed elevated HMGA1 mRNA expression in JAK2V617F but not in mutant calreticulin-driven MPNs; this suggests a contribution of JAK2/STAT5 signaling. The highest HMGA1 mRNA levels appear in AML blasts that harbor EZH2 or ASXL mutations, which indicates that altered transcriptional repression may also be involved. Cytogenetically silent genomic gains of 6p21, including the HMGA1 gene that could lead to its overexpression, have been described in patients with secondary myelodysplastic syndromes (MDSs) and AML rather than MPNs.8 In addition, HMGA1 was identified as part of a leukemic stem cell–associated gene expression signature in MLL-rearranged AML, suggesting that the leukemogenic HMGA1 activity is most likely not limited to MPN progression.9
Activation of HMGA1-controlled transcriptional pathways in matched MF samples after transformation to AML suggests that HMGA1 is a promising therapeutic target. Li et al showed that HMGA1 knockdown increased the antileukemic effects of the ruxolitinib JAK2 inhibitor, which resulted in a significant delay in disease induction upon transplantation of DAMI AML cells into NSG mice. In addition, ruxolitinib treatment reduced BM fibrosis more efficiently in homozygous JAK2V617F;Hmga1+/− mice than in control mice. The fact that a 50% reduction of the Hmga1 gene dose did not affect steady-state hematopoiesis in mice suggests the existence of a therapeutic window. Although several molecules, including DNA minor groove-binding antibiotics, have been shown to functionally interfere with HMGA1, no selective compounds are currently available for clinical use.10 The work by Li et al suggests that both HMGA2 and HMGA1 play a prominent role in the biology of MPNs, particularly in disease progression. More work is needed to comparatively dissect the contributions of both HMGA proteins in MPNs and AML to develop strategies for therapeutic intervention.
Conflict-of-interest disclosure: The author declares no competing financial interests.

