

In this issue of Blood, 1 describe a novel population of triple-positive CD8+ T cells, enriched in the intestinal subcrypt region in gastrointestinal acute graft-versus-host disease (GI-aGVHD), belonging to the Tc1 lineage, and expressing both the retinoic acid receptor α (RARα) and the interleukin-23 receptor (IL-23R) as etiopathologically and therapeutically relevant (see figure).
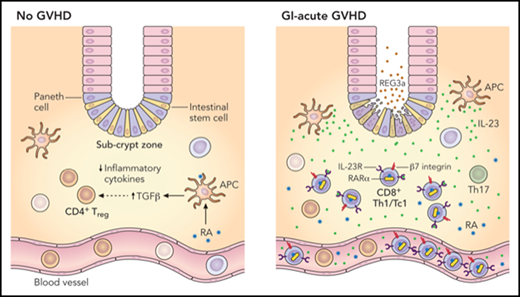
In the normal steady state, RA is involved in the secretion of transforming growth factor β (TGF-β) from antigen-presenting cells (APCs) and promotion of a CD4+ Treg phenotype as well as an overall reduced expression of proinflammatory cytokines. In the setting of GI-aGVHD, infiltration of triple-positive CD8+ T-effector (Teff) cells enriched for Tbet (Th1/Tc1 lineage), RARα, and IL-23R occurs in the intestinal subcrypt zone adjacent to intestinal epithelial stem cells. CD4+ Tregs are also expanded in the subcrypt zone in the setting of GI-aGVHD but to a much lesser extent. Both lineages likely derive from the peripheral blood in response to RA, where (modulated by IL-23/LPS in the setting of GI-aGVHD) expression of GI-trophic receptors (eg, β7 integrin) is induced preferentially on CD8+ Teff cells (vs CD4+ Tregs). Professional illustration by Patrick Lane, ScEYEnce Studios.
In the normal steady state, RA is involved in the secretion of transforming growth factor β (TGF-β) from antigen-presenting cells (APCs) and promotion of a CD4+ Treg phenotype as well as an overall reduced expression of proinflammatory cytokines. In the setting of GI-aGVHD, infiltration of triple-positive CD8+ T-effector (Teff) cells enriched for Tbet (Th1/Tc1 lineage), RARα, and IL-23R occurs in the intestinal subcrypt zone adjacent to intestinal epithelial stem cells. CD4+ Tregs are also expanded in the subcrypt zone in the setting of GI-aGVHD but to a much lesser extent. Both lineages likely derive from the peripheral blood in response to RA, where (modulated by IL-23/LPS in the setting of GI-aGVHD) expression of GI-trophic receptors (eg, β7 integrin) is induced preferentially on CD8+ Teff cells (vs CD4+ Tregs). Professional illustration by Patrick Lane, ScEYEnce Studios.
The lethality of aGVHD is in large part driven by GI involvement.2 A substantial number of patients do not respond to first-line therapy with steroids, and effective second-line therapeutic options remain limited.3 Considerable efforts are underway to optimize the prevention of severe GI-aGVHD while preserving the graft-versus-tumor effect. These efforts critically rely on better understanding the pathophysiology of aGVHD, and in particular the mediators of GI tissue–specific epithelial injury.
By using an immunohistochemistry approach on archival GI biopsy specimens to characterize retinoic acid (RA)–responsive immune cells localized to the subcrypt zone of the upper and lower GI tract (adjacent to intestinal epithelial stem cells), Ball et al found that RARα-expressing CD8+ T cells were enriched in patients with GI-aGVHD. The frequency of this cell population in the lower GI tract helped distinguish patients with greater clinical and histopathologic severity of GI-aGVHD and GVHD-associated mortality.
RA is a known mediator of the homing of certain subsets of T cells to the GI tract. As a potentiator of T-cell activity, the role of RA in aGVHD previously seemed both important and ambiguous in part because of its dependence on the microenvironmental context. A mouse model of aGVHD showed that in the absence of RA-mediated signaling, specifically via depletion of RARα in T cells, the development of lethal GI-aGVHD was attenuated.4,5 However, in a physiological context, RA is associated with an increase in anti-inflammatory regulatory T cells (Tregs) and a concurrent decrease in the secretion of inflammatory cytokines.6 Conversely, in an inflammatory milieu, RA is associated with the expansion of effector Th1 and Th17 T cells, phenotypes that can potentiate tissue damage.5
Ball et al have found that although both RA-responsive CD8+ T and CD4+ Tregs were increased in the subcrypt zone of patients with GI-aGVHD, the infiltration of CD8+ T cells was significantly greater. The authors explored this selective enrichment of CD8+ T cells in vitro with exogenous administration of RA, and they found that even though RA induced an upregulation of GI-trophic receptors in both CD8+ and CD4+ Tregs, at higher doses the proliferation of CD4+ Tregs was suppressed. Furthermore, the authors found that IL-23 significantly reduced the RA-induced expansion of CD4+ Tregs, which accounts for selective fivefold expansion of the GI-trophic CD8+ T-cell subset with a high coexpression of RARα, IL-23R, and Tc1 lineage markers within the IL-23–rich GI-aGVHD subcrypt zone. These triple-positive RA-responsive CD8+ T cells were also increased in the peripheral blood of GI-aGVHD patients, but not in those with non-GI-aGVHD or in allogeneic stem cell transplant recipients without GVHD.
This elegant study by Ball et al identifies a link between RA and an effector GI-trophic CD8+ T-cell population that seems capable of inducing intestinal stem cell injury (the latter is inferred, not demonstrated). Because specific blockade of RARα abrogated the expansion of these triple-positive CD8+ T cells, a resulting model has IL-23 modulating the effect of RA on T cells in GI-aGVHD, favoring the expansion and GI-tropism of the triple-positive effector CD8+ subset while suppressing the expansion of CD4+ Tregs.
Previous studies have shown that initiation of aGVHD is dependent on donor Th1 T cells expressing IL-23R.7 Furthermore, secretion of IL-23 by colonic resident antigen-presenting cells after conditioning chemotherapy is a potent inducer of GI-aGVHD.8 Blockade of IL-23 signaling with ustekinumab in combination with tacrolimus and sirolimus in a recent clinical trial resulted in a lower serum concentration of REG3a, a cytokine associated with increased severity of GI-aGVHD.9 Although the trial was not powered to assess clinical outcomes, it highlighted the need for further study of the mediators of IL-23 signaling in aGVHD and specifically for the elucidation of effector T cells involved in IL-23–driven tissue destruction.
This study suggests that although a proinflammatory environment rich in IL-23 can induce CD4+ T cells to initiate aGVHD,7 further propagation of tissue damage in the presence of RA may require CD8+ Tc1 cells. Interestingly, the authors did not observe an IL-23–mediated expansion of Th17 cells in GI-aGVHD, somewhat counter to observations from previous studies.8 Also notable was that expansion of RA-responsive cells was not correlated with IL-33 receptor expression in the intestinal subcrypts, suggesting that RA-mediated GI-aGVHD effects may be independent of the dysregulation of IL-33/ST2 signaling known to be relevant in GVHD.10 In addition, the authors found that lipopolysaccharide (LPS), a known mediator of aGVHD, in combination with RA had a larger effect on the expansion of triple-positive CD8+ T cells than the combination of IL-23 and RA. Future studies of RA-mediated GI-aGVHD will likely require dissection of both IL-23– and LPS-mediated downstream signaling.
This work provides a reasonable rationale for investigating RA-responsive triple-positive CD8+ T cells and RA-mediated signaling as targets for therapeutic intervention in GI-aGVHD. The study by Ball et al documented that RARα blockade prevented the expansion of the triple-positive CD8+ T cells in vitro, but it remains to be determined whether this would be a viable therapeutic strategy. Alternative approaches suppressing the intestinal epithelial stem cell injury induced by the subcrypt CD8+ T-cell subset would be worth investigating. Whether any of these approaches can augment the efficacy of therapeutics like vedolizumab (clinicaltrials.gov #NCT03657160) that interdict the GI tropism of T cells also remains to be seen.
Conflict-of-interest disclosure: J.K. has served as a consultant for EMD Serono, Merck, GentiBio, Biolojic Design, Equillium, and Moderna Therapeutics; has served on an advisory board for Cugene and Therakos; and has received research support from Equillium, Amgen, Bristol Myers Squibb, Miltenyi Biotec, Regeneron, and Clinigen. R.M.S. declares no competing financial interests.

