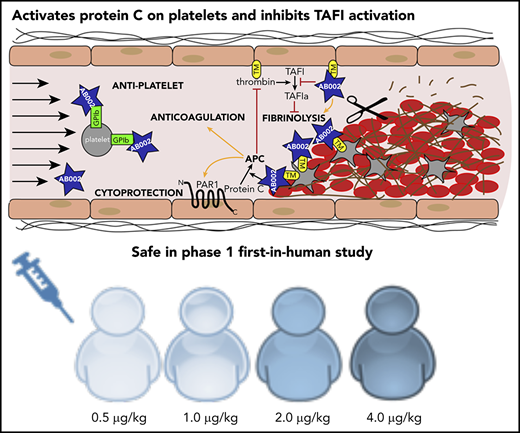
Key Points
AB002 (E-WE thrombin) rapidly interrupts thrombus development and prevents acute vascular graft occlusion in baboons.
In a phase 1 first-in-human clinical trial, AB002 produced transient protein C activation without substantial systemic anticoagulation.

Abstract
Although thrombin is a key enzyme in the coagulation cascade and is required for both normal hemostasis and pathologic thrombogenesis, it also participates in its own negative feedback via activation of protein C, which downregulates thrombin generation by enzymatically inactivating factors Va and VIIIa. Our group and others have previously shown that thrombin’s procoagulant and anticoagulant activities can be effectively disassociated to varying extents through site-directed mutagenesis. The thrombin mutant W215A/E217A (WE thrombin) has been one of the best characterized constructs with selective activity toward protein C. Although animal studies have demonstrated that WE thrombin acts as an anticoagulant through activated protein C (APC) generation, the observed limited systemic anticoagulation does not fully explain the antithrombotic potency of this or other thrombin mutants. AB002 (E-WE thrombin) is an investigational protein C activator thrombin analog in phase 2 clinical development (clinicaltrials.gov NCT03963895). Here, we demonstrate that this molecule is a potent enzyme that is able to rapidly interrupt arterial-type thrombus propagation at exceedingly low doses (<2 µg/kg, IV), yet without substantial systemic anticoagulation in baboons. We demonstrate that AB002 produces APC on platelet aggregates and competitively inhibits thrombin-activatable fibrinolysis inhibitor (carboxypeptidase B2) activation in vitro, which may contribute to the observed in vivo efficacy. We also describe its safety and activity in a phase 1 first-in-human clinical trial. Together, these results support further clinical evaluation of AB002 as a potentially safe and effective new approach for treating or preventing acute thrombotic and thromboembolic conditions. This trial was registered at www.clinicaltrials.gov as #NCT03453060.
Introduction
Currently approved antithrombotic and thrombolytic therapies are effective at halting and reversing thrombosis and thromboembolism; however, the efficacy of current treatments can be offset by their bleeding side effects. As a result, in many cases, antithrombotic drugs such as heparins or plasminogen activators cannot be dosed to their full efficacy. Accordingly, there remains a need for new acute use antithrombotics that are effective while being hemostatically safe.
Similar to tissue plasminogen activator (tPA)-induced plasmin, thrombin-induced activated protein C (APC) is an endogenous antithrombotic enzyme. APC anticoagulates blood by inhibiting thrombin generation through enzymatic degradation of coagulation cofactors Va and VIIIa. Infused APC is a potent systemic anticoagulant at antithrombotic doses in primates,1,2 and improves neurological outcomes of experimental stroke in mice.3,4 Beyond its anticoagulant activity, APC activates cytoprotective mechanisms through protease-activated receptor 1 (PAR1)-mediated signaling in endothelial cells,4-6 reduces prothrombotic and proinflammatory neutrophil extracellular trap formation (NETosis),7 and helps maintain the integrity of the blood-brain barrier.8 However, systemic APC administration has the capacity to impair hemostatic thrombin generation, as APC that is not surface- or receptor-bound remains an active anticoagulant in the fluid phase of blood. Although potentially effective, the use of recombinant APC (drotrecogin alfa; Lilly) for treating thrombosis has not been clinically pursued.
Apart from snake venoms,9 thrombomodulin (TM)-bound thrombin is the only known physiologically relevant protein C activator enzyme.10-12 We have shown that low-dose thrombin infusion (1 U/kg per minute ≈ 0.4 µg/kg per minute) is antithrombotic in our baboon vascular graft thrombosis model through endogenous APC generation,13 but the therapeutic window of wild-type thrombin is far too narrow for its safe clinical utilization. Structural analyses, alanine scanning, and other studies identified key residues involved in thrombin’s substrate specificity, leading to the rational design of thrombin analogs with impaired procoagulant activity.14-20 Our original thrombin mutant W215A/E217A (WE thrombin) has hundred-fold to several thousand-fold reduced catalytic activity toward its prothrombotic substrates, including fibrinogen and platelet PAR1, but retains activity toward the antithrombotic substrate protein C when in complex with TM.14 In baboons, low-dose WE thrombin prevented thrombus formation comparable to interventional IV doses of low-molecular-weight heparin or high-dose exogenous APC, but without detectable primary hemostasis impairment.16 Also, WE thrombin significantly improved the outcome in a murine ischemic stroke model without increased bleeding.21 Surprisingly, the WE thrombin-induced antithrombotic effect has been observed at a much lower level of systemic anticoagulation than what was needed to achieve a similar efficacy when exogenous APC infusion was used in our baboon thrombosis model.1,16
Because there is no obvious explanation for the potency of WE thrombin at low levels of anticoagulation, we hypothesized that one mechanism by which WE thrombin could exert its antithrombotic activity was through protein C activation under intravascular shear flow on the vessel wall or thrombus surface.16 In this proposed model, treatment with WE thrombin exploits an endogenous targeting system via cell-associated thrombin receptors such as platelet glycoprotein Ib (GPIb),22,23 thereby delivering WE thrombin directly to the accumulating thrombus, which is the site of desired pharmacological activity.16 We proposed that at the blood thrombus interface, surface-associated endogenous APC is generated by WE thrombin, in situ, with a slow or limited escape of APC into the circulation, causing only moderate and transient systemic anticoagulation. Indeed, we previously confirmed that WE thrombin accumulates in developing experimental thrombi.16 However, the mechanism of APC generation on the thrombus surface spatially distant from the endothelium remained obscure. We suspected that WE thrombin may also compete with thrombin for GPIb binding,22 and these mechanisms may be at least partially responsible for downregulating thrombin generation and platelet activation on thrombi, and preventing thrombogenesis.
AB002 (E-WE thrombin) is an Escherichia coli–derived protein C activator recombinant thrombin analog that is in development for thrombotic and inflammatory indications. Although the antithrombotic and anti-inflammatory properties of the mammalian cell-derived recombinant WE thrombin have been well documented in disease prevention models,15,16,21,24,25 the E coli–produced analog AB002 has yet to be fully described. Therefore, we sought to characterize the molecular properties of AB002, evaluate its effectiveness using a new acute thrombosis treatment model in baboons, and identify additional molecular mechanisms that may help explain its potent antithrombotic activities. Based upon these compelling proof-of-principle efficacy studies, we further explored AB002’s safety and activity in a phase 1 clinical trial in healthy adult subjects.
Methods
AB002 manufacturing
E coli–expressing prethrombin-2 containing the W215A/E217A active site mutations was generated by using site-directed mutagenesis and an established E coli expression system as previously described.26 The prethrombin-2 zymogen was then activated using ecarin (Pentapharm). More details are provided in supplemental Methods (available on the Blood Web site).
Activity of AB002
As a bridge to developing a product-specific potency standard, thrombin standard lot K (World Health Organization [WHO] 2nd International Standard 01/580) was used as the primary reference standard for testing specific enzymatic potency of AB002. Chromogenic substrate (S-2238; Chromogenix) cleavage was monitored as an increase in absorbance at 405 nm. The rate of substrate hydrolysis by AB002 was used to measure the potency relative to that of a WHO thrombin standard by parallel line analysis (logarithmic response). The relative potency of the AB002 samples against the WHO thrombin standard were determined using the average slope values. The specific activity determined for AB002 is not indicative of clotting activity. To evaluate the activity of AB002 against select thrombin substrates in vitro, AB002 was incubated with several physiological or synthetic thrombin substrates.14 Details are provided in supplemental Methods.
Pharmacodynamics of AB002 in baboons
Details are provided in supplemental Methods.
Acute thrombosis treatment models
Vascular graft thrombosis treatment experiments were performed using 12 juvenile male baboons (Papio anubis, 10-14 kg) with chronic exteriorized femoral arteriovenous shunts, as described elsewhere.13,27 All baboon experiments were approved by the Institutional Animal Care and Use Committee of Oregon Health & Science University.
Expanded polytetrafluoroethylene grafts were coated with collagen as previously described for our thrombosis prevention model.27 Graft length was 20 mm with an internal diameter (i.d.) of either 2 or 4 mm. The grafts were deployed into arteriovenous shunts for up to 90 minutes. The flow rate was maintained at 100 ± 10 mL/min via distal shunt clamping. Experiments were stopped if the unclamped flow rate dropped below 20 mL/min, signaling imminent occlusion.
For experiments using 4-mm i.d. grafts, AB002 (1.25, 1.5, and 2 µg/kg; IV bolus), tPA (alteplase [Genentech], 1 mg/kg; IV as 1/3 bolus and 2/3 infusion over 30 minutes), enoxaparin (Sanofi-Aventis, 2 mg/kg; IV bolus), or APC (American Red Cross, 25 and 50 µg/kg; IV bolus) was administered 30 minutes after thrombus initiation. Control animals received either saline or no treatment. The experiment was continued for an additional 60 minutes to monitor thrombus propagation. For experiments utilizing 2-mm i.d. grafts, AB002 (2.5, 5.0, and 10 µg/kg; IV bolus) or saline was administered 15 minutes after thrombus initiation. The experiment was continued for an additional 45 minutes or until graft occlusion.
Thrombus formation was assessed by real-time dynamic γ camera imaging (GE-400T or a GE-Brivo NM615; GE Healthcare) of 111In-radiolabeled platelets, and further assessed by measuring end point 125I-labeled fibrinogen/fibrin deposition. Platelet accumulation data were collected and analyzed as described previously.13,27
APC generation on platelet aggregates in vitro
APC generation by AB002 on collagen-activated washed human platelets was measured by HAPC 1555 enzyme-linked immunosorbent assay (ELISA). Details are provided in supplemental Methods.
TAFI activation assays
Activation of thrombin-activatable fibrinolysis inhibitor (TAFI; carboxypeptidase B2) was evaluated using a capillary-based immunodetection system (Wes from ProteinSimple) and by colorimetric assay. Details are provided in supplemental Methods.
In vitro fibrinolysis experiments
Standard activated partial thromboplastin time (aPTT) reactions were performed using HemosIL reagent (Instrument Laboratories) and a KC-4 coagulometer. Briefly, 40 µL of healthy (NIBSC SSCLOT4) or TAFI-deficient (Affinity Biologicals) plasma was incubated with 40 µL of aPTT reagent for 180 seconds. At 120 seconds into the incubation, tPA (40 µg/mL) alone or tPA and AB002 (200 µg/mL) were added at a 1/20 dilution (for both) to 20 mM CaCl2. At this concentration of AB002, TM is the limiting factor because no exogenous TM was added to the reaction and cell-free soluble TM in plasma is reportedly in the range of 0.2 to 5 nM.28,29 At the end of the incubation, 40 µL of this CaCl2 solution was added to the reaction. Lysis time was recorded as the time starting immediately after clot formation until the clot was lysed (ie, the time it takes for the clot to liquefy and the metal sphere in the KC-4 vial to stop rotating).
AB002 pharmacology evaluation during cynomolgus monkey toxicology studies
Details are provided in supplemental Methods.
Phase 1 first-in-human study
A phase 1, randomized, double-blind, placebo-controlled, single-ascending dose study was conducted in healthy adult volunteers at a single site (Celerion, Tempe, AZ). The protocol (clinicaltrials.gov NCT03453060) was approved by the Advarra Institutional Review Board in accordance with the Declaration of Helsinki and International Conference on Harmonisation (ICH) Good Clinical Practice. All subjects provided written informed consent.
The study consisted of 4 ascending dose cohorts (0.5, 1.0, 2.0, 4.0 µg/kg) in which subjects were randomized to receive a single IV bolus of AB002 (n = 4 per cohort) or placebo (n = 1 per cohort, except for the 0.5 µg/kg dose level, in which n = 2). Both investigators and subjects were blinded to the treatment and a data safety monitoring committee provided oversight. The primary study end point was the number and severity of treatment-emergent adverse events (TEAEs). The secondary study end point was the magnitude and duration of plasma APC–protein C inhibitor (APC-PCI) complexes following drug administration. APC-PCI was measured using a commercial ELISA method (Bioporto Diagnostics, Hellerup, Denmark) and served as a surrogate biomarker for drug exposure. Protein C levels were measured in plasma using a commercial competitive ELISA kit per the manufacturer’s instructions (Innovative Research, Inc). D-dimer levels were measured in plasma using a commercial ELISA kit per the manufacturer’s instructions (Sekisui Diagnostics, LLC). Prothrombin fragment 1.2 levels were measured in plasma using a commercial ELISA kit per the manufacturer’s instructions (US Biological). Immunogenicity was evaluated at Haemtech Biopharma Services using bridging-type ELISAs for the detection and titer of any potential antidrug antibodies or anti-human wild-type (WT) thrombin antibodies. Detailed descriptions of these assays can be found in the supplemental Methods.
Statistics
Statistical analysis was performed using SigmaPlot 11.2 or GraphPad Prism 5. A P value of <.05 was considered statistically significant for all tests.
Results
AB002 potency, thrombin substrate specificity, and pharmacological activity
Our early stage stability and lot release potency assay, based on direct chromogenic substrate cleavage, found the 8 independent manufacturing lots of AB002 to have a specific potency of 32 ± 3 U/mg (mean plus or minus standard error of the mean [SEM]), where WHO thrombin was used as an interim reference standard for amidolytic activity. The substrate specificity of W215A/E217A mutant thrombin produced in a mammalian expression system (baby hamster kidney cells) has previously been described.14,30 We repeated these experiments using several AB002 manufacturing lots. We also measured the plasma-clotting potential of AB002, and tested its pharmacological activity in baboons by measuring plasma APC-PCI concentrations as a biomarker for APC generation. These experiments confirmed that the E coli–produced WE thrombin had a comparable activity profile to the mammalian prototype with no measurable fibrin-clotting potential (supplemental Table 1) and is pharmacologically active in baboons (supplemental Figure 1). WE thrombin has been shown previously to not directly activate platelets,22 and we now confirm that AB002 also does not alter thrombin-induced platelet aggregation (data not shown).
AB002 interrupts experimental thrombus propagation in baboons
To mimic a large-vessel acute thrombotic event, where the platelet-rich thrombi are in a rapid propagation phase, AB002, tPA, or enoxaparin was administered 30 minutes after deploying a 4-mm i.d. collagen-coated vascular graft in baboons. By 30 minutes, accumulation within the graft segments averaged 1.6 × 109 ± 0.1 × 109 platelets and was not statistically different between groups. Platelet accumulation within the thrombus tail region (distal to the graft) that extended up to 10 cm downstream of the graft segments averaged 1.2 × 109 ± 0.1 × 109 for all groups at 30 minutes. Although the control animals showed robust thrombus propagation, AB002 rapidly interrupted both graft and tail platelet accumulation within 10 to 20 minutes (Figure 1A-B,E), which was similar to the results obtained after high-dose enoxaparin administration. Administration of tPA interrupted and began to reverse platelet accumulation within the collagen-coated graft segment only after ∼50 minutes, whereas tail platelet accumulation was reversed within 10 minutes of tPA dosing. End-point fibrin accumulation was significantly reduced in the tPA-treated group (0.57 ± 0.19 mg), the enoxaparin-treated group (0.44 ± 0.08 mg), and in the 2 µg/kg AB002-treated group (0.72 ± 0.03 mg) compared with controls (1.33 ± 0.17 mg; Figure 1C), whereas the other dose levels of AB002 trended lower but did not reach statistical significance.
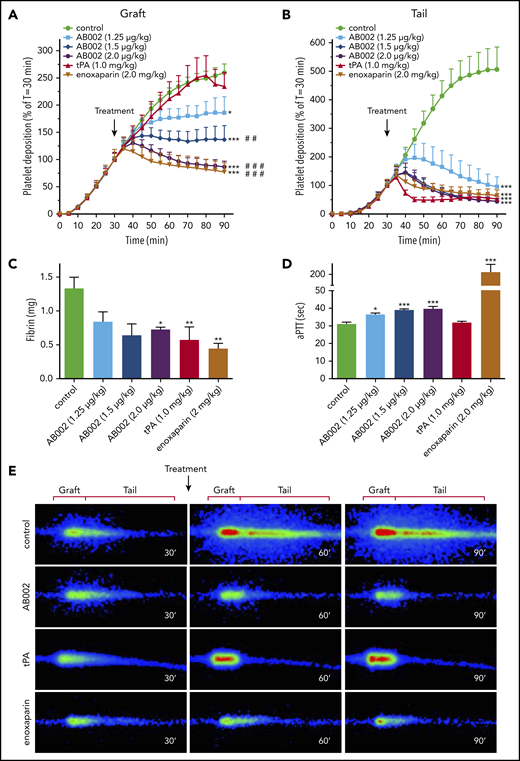
AB002 interrupts thrombus formation in a primate thrombosis model. (A-B) Real-time platelet accumulation within collagen-coated synthetic vascular grafts (4-mm i.d.; 20 mm length) (A) and the tail region where the thrombus elongated distal to the graft (B). These devices were temporarily inserted into the femoral arteriovenous shunt of juvenile baboons. Blood flow through the device was adjusted to 100 mL/min, producing an initial shear rate of 265 s−1. Thrombi were allowed to grow for 30 minutes prior to intervention (black arrow). The average thrombus size at the time of intervention was set to 100%. Control experiments (n = 15) received no intervention or saline, whereas treatments were either tPA (n = 8) or increasing doses of E-WE thrombin (n = 4-7 per group). Posttreatment platelet accumulation was monitored for an additional 60 minutes. (C) Total fibrin deposition within the grafts was assessed at the end of each experiment. (D) aPTT was measured in platelet-poor plasma samples collected 10 minutes posttreatment. (E) Real-time γ-camera images of the developing thrombi taken at 30, 60, and 90 minutes. Images were recorded at 5-minute intervals and quantified in panels A and B. Each row of images shows representative images at each time point for control, AB002- (2 µg/mL), tPA-, and enoxaparin-treated baboons. Dimensions used to quantify graft and tail thrombus are indicated above each column. All data are expressed as mean plus or minus SEM. Asterisks denote significant differences to control (*P < .05; **P < .01; ***P < .001), pound symbols indicate significant differences to tPA (#P < .05; ##P < .01; ###P < .001). For panel B, all treatment groups, including tPA, are significantly different from control (P < .001), but not different from each other. Real-time platelet deposition was analyzed by 2-way ANOVA, and terminal fibrin and aPTT were analyzed using 1-way ANOVA.
AB002 interrupts thrombus formation in a primate thrombosis model. (A-B) Real-time platelet accumulation within collagen-coated synthetic vascular grafts (4-mm i.d.; 20 mm length) (A) and the tail region where the thrombus elongated distal to the graft (B). These devices were temporarily inserted into the femoral arteriovenous shunt of juvenile baboons. Blood flow through the device was adjusted to 100 mL/min, producing an initial shear rate of 265 s−1. Thrombi were allowed to grow for 30 minutes prior to intervention (black arrow). The average thrombus size at the time of intervention was set to 100%. Control experiments (n = 15) received no intervention or saline, whereas treatments were either tPA (n = 8) or increasing doses of E-WE thrombin (n = 4-7 per group). Posttreatment platelet accumulation was monitored for an additional 60 minutes. (C) Total fibrin deposition within the grafts was assessed at the end of each experiment. (D) aPTT was measured in platelet-poor plasma samples collected 10 minutes posttreatment. (E) Real-time γ-camera images of the developing thrombi taken at 30, 60, and 90 minutes. Images were recorded at 5-minute intervals and quantified in panels A and B. Each row of images shows representative images at each time point for control, AB002- (2 µg/mL), tPA-, and enoxaparin-treated baboons. Dimensions used to quantify graft and tail thrombus are indicated above each column. All data are expressed as mean plus or minus SEM. Asterisks denote significant differences to control (*P < .05; **P < .01; ***P < .001), pound symbols indicate significant differences to tPA (#P < .05; ##P < .01; ###P < .001). For panel B, all treatment groups, including tPA, are significantly different from control (P < .001), but not different from each other. Real-time platelet deposition was analyzed by 2-way ANOVA, and terminal fibrin and aPTT were analyzed using 1-way ANOVA.
All doses of AB002 increased the aPTT compared with baseline in a dose-dependent manner (Figure 1D), with a maximum prolongation of only 1.3-fold above baseline values at the most effective dose (2 µg/kg). Consistent with its mechanism of action, tPA did not prolong the aPTT, whereas high-dose IV enoxaparin increased the aPTT approximately sevenfold above baseline.
In a single baboon, an IV bolus of APC (25 µg/kg or 50 µg/kg, n = 1 each) was administered at 30 minutes using the same thrombosis model (supplemental Figure 2). APC, while increasing the aPTT 1.4-fold to 1.6-fold, did not robustly influence platelet accumulation on the graft or tail, but did appear to decrease graft-associated fibrin deposition compared with control values.
AB002 interrupts occlusive thrombus formation in baboons
To mimic a high-shear small-vessel occlusive thrombotic event, we administered AB002 15 minutes after deploying a 2-mm i.d. collagen-coated vascular graft in baboons. Figure 2A shows real-time platelet accumulation in the whole thrombus. In control experiments, the thrombus grew steadily until each graft occluded, with an average occlusion time of 29 ± 2 minutes. After AB002 administration, the thrombus continued growing for 5 to 10 minutes before growth halted and “thrombolysis” (or platelet disaggregation) began. The baseline occlusion time ranged from 21 to 33 minutes (Figure 2B). Intervention with AB002 prolonged time to occlusion at all doses, and the graft did not occlude in 67% of the AB002-treated animals compared with 100% occlusion in controls. There was no obvious additional benefit of increasing the AB002 dose beyond 2.5 µg/kg. Although there does appear to be a dose saturation effect, we are reluctant to conclude that there is a detrimental effect on efficacy at higher doses. We propose that this observation may be related to the limited number of animals studied.
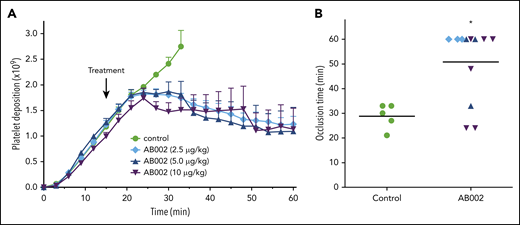
AB002 interrupts occlusive thrombus formation. (A) Real-time platelet accumulation within small diameter collagen-coated synthetic vascular grafts (2-mm i.d.; 20 mm length). Blood flow through the device was adjusted to 100 mL/min, producing an initial wall shear rate of 2120 s−1. Thrombi were allowed to grow for 15 minutes before treatment (black arrow). E-WE thrombin was given as an IV bolus (n = 3-6 per treatment group); controls (n = 5) received no treatment. Data are means plus or minus SEM. (B) Time to occlusion is shown. Horizontal bars denote mean of all data points. All control grafts occluded within 21 to 33 minutes. AB002 treatment significantly increased the time to occlusion (*P < .05; Gehan-Breslow-Wilcoxon test), with 8 of 12 grafts remaining patent for the entire 60 minutes.
AB002 interrupts occlusive thrombus formation. (A) Real-time platelet accumulation within small diameter collagen-coated synthetic vascular grafts (2-mm i.d.; 20 mm length). Blood flow through the device was adjusted to 100 mL/min, producing an initial wall shear rate of 2120 s−1. Thrombi were allowed to grow for 15 minutes before treatment (black arrow). E-WE thrombin was given as an IV bolus (n = 3-6 per treatment group); controls (n = 5) received no treatment. Data are means plus or minus SEM. (B) Time to occlusion is shown. Horizontal bars denote mean of all data points. All control grafts occluded within 21 to 33 minutes. AB002 treatment significantly increased the time to occlusion (*P < .05; Gehan-Breslow-Wilcoxon test), with 8 of 12 grafts remaining patent for the entire 60 minutes.
AB002 generates APC on activated platelet surfaces and concentration-dependently inhibits TAFI activation in vitro
Because the observed AB002-induced aPTT prolongation was only marginal, and consequently circulating APC did not fully explain the observed robust antithrombotic effect, we examined whether AB002 could generate localized APC independent of the endothelium. We used collagen-stimulated human platelet aggregates as a surface for protein C activation. Figure 3A shows that platelets and AB002 can combine to promote conversion of protein C to APC. The active site serine mutant S195A-WE thrombin did not generate APC, demonstrating that this conversion depends upon a functional active site. WT thrombin was previously shown to activate protein C in a TM-dependent manner in the presence of thrombin-stimulated platelets.28 We now demonstrate that AB002’s ability to generate APC in the presence of platelet aggregates also depends on TM, as indicated by blocking experiments (Figure 3B). Recombinant receptor-associated protein, which inhibits ligand binding to low-density lipoprotein receptors and inhibits protein C binding to platelets,31 also significantly reduced AB002-mediated APC generation.
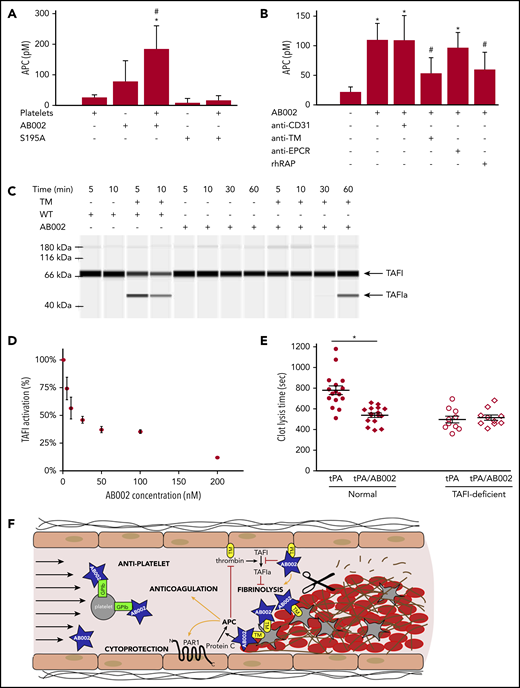
AB002 catalytically converts protein C to APC on activated platelet aggregates and inhibits thrombin-mediated TAFI activation. (A) Washed human platelets were activated and aggregated by collagen and treated with 50 nM AB002 or S195A-WE and 100 nM protein C. Platelets treated with protein C only and thrombin mutants incubated with protein C in the absence of platelets were prepared at the same time. Supernatants were collected after 1 hour and APC measured. Data are shown as the mean ± 1 standard deviation for 4 independent experiments (n = 4 donors). *P < .05 compared with platelet-only control and #P < .05 compared with platelet-free AB002-only control (repeated measures ANOVA and Holm-Sidak 2-tailed post hoc tests). (B) Washed human platelets were activated by collagen and pretreated with blocking reagents for 1 hour: 40 µg/mL anti-TM or anti-endothelial protein C receptor (EPCR), or 20 µg/mL recombinant human receptor-associated protein (rhRAP). Anti-CD31 (40 µg/mL) was used as a negative control for antibody blocking. Pretreated platelets were incubated with 50 nM AB002 and 100 nM protein C or protein C alone, as indicated. Supernatants were collected after 1 hour and APC measured. Data are shown as the mean ± 1 standard deviation for 4 independent experiments (n = 4 donors). *P < .05 compared with platelet-only control and #P < .05 compared with platelets with AB002 control (repeated measures ANOVA and Holm-Sidak 2-tailed post hoc tests). (C) WT α-thrombin (WT) or AB002 were added to HEPES buffered saline (HBS) with or without TM, purified TAFI was added, and samples incubated at room temperature for the indicated times before evaluating the activation of TAFI. Samples containing 500 nM purified TAFI and 150 nM WT with or without 15 nM TM (lanes 1-4) or 150 nM AB002 with or without 15 nM TM (lanes 5-12) were subjected to automated western blot analysis. (D) AB002 was added to 5 nM WT and 5 nM TM in increasing concentrations from 5 to 200 nM and TAFI activity measured. The activity of WT in the absence of AB002 was set to 100%. (E) A standard aPTT reaction was performed in a KC4 coagulometer using either healthy or TAFI-deficient platelet-poor plasma; at the end of the incubation time, 2 µg/mL tPA with or without 10 µg/mL AB002 was added, and the reaction was recalcified to initiate clot formation. aPTT was measured and time to complete lysis of the clot was monitored. Horizontal bars denote mean of all data points plus or minus SEM. *P < .05 (independent Student t test). (F) Putative mechanism by which low doses of AB002 can rapidly interrupt developing thrombi through its ability to locally activate protein C on the platelet-rich thrombus luminal surface and by competitive inhibition of TAFI activation (TAFIa).
AB002 catalytically converts protein C to APC on activated platelet aggregates and inhibits thrombin-mediated TAFI activation. (A) Washed human platelets were activated and aggregated by collagen and treated with 50 nM AB002 or S195A-WE and 100 nM protein C. Platelets treated with protein C only and thrombin mutants incubated with protein C in the absence of platelets were prepared at the same time. Supernatants were collected after 1 hour and APC measured. Data are shown as the mean ± 1 standard deviation for 4 independent experiments (n = 4 donors). *P < .05 compared with platelet-only control and #P < .05 compared with platelet-free AB002-only control (repeated measures ANOVA and Holm-Sidak 2-tailed post hoc tests). (B) Washed human platelets were activated by collagen and pretreated with blocking reagents for 1 hour: 40 µg/mL anti-TM or anti-endothelial protein C receptor (EPCR), or 20 µg/mL recombinant human receptor-associated protein (rhRAP). Anti-CD31 (40 µg/mL) was used as a negative control for antibody blocking. Pretreated platelets were incubated with 50 nM AB002 and 100 nM protein C or protein C alone, as indicated. Supernatants were collected after 1 hour and APC measured. Data are shown as the mean ± 1 standard deviation for 4 independent experiments (n = 4 donors). *P < .05 compared with platelet-only control and #P < .05 compared with platelets with AB002 control (repeated measures ANOVA and Holm-Sidak 2-tailed post hoc tests). (C) WT α-thrombin (WT) or AB002 were added to HEPES buffered saline (HBS) with or without TM, purified TAFI was added, and samples incubated at room temperature for the indicated times before evaluating the activation of TAFI. Samples containing 500 nM purified TAFI and 150 nM WT with or without 15 nM TM (lanes 1-4) or 150 nM AB002 with or without 15 nM TM (lanes 5-12) were subjected to automated western blot analysis. (D) AB002 was added to 5 nM WT and 5 nM TM in increasing concentrations from 5 to 200 nM and TAFI activity measured. The activity of WT in the absence of AB002 was set to 100%. (E) A standard aPTT reaction was performed in a KC4 coagulometer using either healthy or TAFI-deficient platelet-poor plasma; at the end of the incubation time, 2 µg/mL tPA with or without 10 µg/mL AB002 was added, and the reaction was recalcified to initiate clot formation. aPTT was measured and time to complete lysis of the clot was monitored. Horizontal bars denote mean of all data points plus or minus SEM. *P < .05 (independent Student t test). (F) Putative mechanism by which low doses of AB002 can rapidly interrupt developing thrombi through its ability to locally activate protein C on the platelet-rich thrombus luminal surface and by competitive inhibition of TAFI activation (TAFIa).
Because WT thrombin can inhibit fibrinolysis through TAFI activation, we investigated whether AB002 could act as a competitive antagonist to TAFI activation and, in turn, promote fibrinolysis. We found AB002 to be a poor TAFI activator compared with WT thrombin, even in the presence of TM (Figure 3C; supplemental Figure 3). Quantitative assays indicated that AB002 is able to concentration-dependently inhibit thrombin/TM-mediated TAFI activation (Figure 3D). Consequently, we tested whether AB002 could aid in fibrinolysis. As shown in Figure 3E, AB002 significantly reduced tPA-mediated clot lysis time, from 780 ± 40 seconds to 540 ± 20 seconds, an effect that was not observed in TAFI-deficient plasma. These data taken together suggest a putative mechanism whereby AB002 can promote fibrinolysis and locally activate protein C at the thrombus interface (Figure 3F). Although AB002 reduced TAFI activation in vitro, a western blot analysis was not sensitive enough to confirm a systemic reduction of TAFI activation in baboons treated with AB002 vs control during the thrombosis experiments (data not shown).
AB002 generates APC in cynomolgus monkeys with no observed toxicity
In baboons, a pharmacokinetic (PK) assay was only sensitive to detect AB002 when administered well over the intended (supratherapeutic) doses (≥10 µg/kg), likely due to receptor binding and rapid clearance from circulation. In toxicity studies with supratherapeutic doses given to cynomolgus monkeys, plasma AB002 concentrations were detectable at early time points, peaking at 15 minutes postadministration and returning quickly to baseline (supplemental Figure 4A). As a marker for APC generation, the APC-PCI complex was evaluated in plasma. Elevated APC-PCI levels ranged from a minimum of 250 ng/mL APC-PCI when AB002 was administered at 12.5 µg/kg to a maximum of 800 ng/mL at 50 µg/kg (supplemental Figure 4B). The observed pharmacodynamic (PD) profile for protein C activation closely followed the PK profile for plasma concentrations of AB002.
The anticipated PD effect of mild-to-moderate aPTT prolongation caused by some APC released into systemic circulation occurred immediately postdose and persisted for at least 1 hour (supplemental Figure 4C). Prolongation of aPTT was observed to be dose-dependent, with peak prolongation over baseline values ranging from 2.2-fold to 3.2-fold at the highest doses of AB002. The temporal aPTT prolongation corresponded similarly to the profile of plasma APC-PCI concentration. The study found no signs of toxicity associated with any dose of AB002.
First-in-human study
In this phase 1 study, 21 subjects were enrolled, received the study medication, completed the study per protocol, and were included in the safety and PD analyses. AB002 was given IV to 16 healthy volunteers at doses ranging from 0.5 µg/kg to 4 µg/kg with an additional 5 subjects receiving placebo. The dose-escalation schedule is shown in Figure 4, and subject demographics are shown in Table 1.
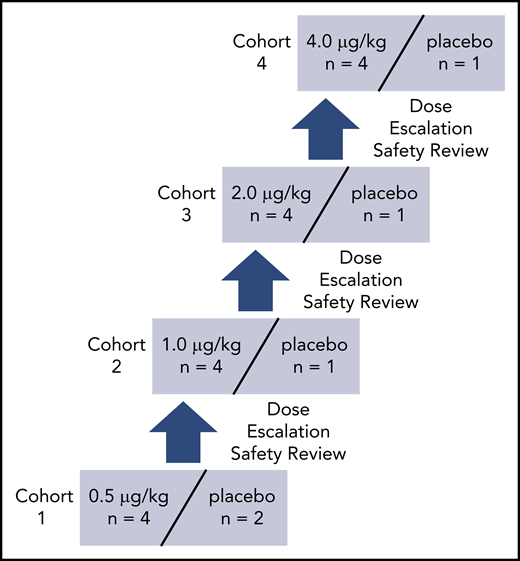
Dose-escalation schedule for AB002 phase 1 clinical study. Subjects were screened and those who met the eligibility criteria were randomized to receive either AB002 or placebo at a 4:1 ratio, except for cohort 1, in which the ratio was 4:2 to include 2 sentinel subjects (1 active and 1 placebo). Each cohort was dosed sequentially. Prior to dose escalation, all safety data were collected from each subject through day 14 and reviewed by the safety review committee during a dose-escalation safety review meeting.
Dose-escalation schedule for AB002 phase 1 clinical study. Subjects were screened and those who met the eligibility criteria were randomized to receive either AB002 or placebo at a 4:1 ratio, except for cohort 1, in which the ratio was 4:2 to include 2 sentinel subjects (1 active and 1 placebo). Each cohort was dosed sequentially. Prior to dose escalation, all safety data were collected from each subject through day 14 and reviewed by the safety review committee during a dose-escalation safety review meeting.
There were no serious adverse events experienced and no subjects were removed from the study due to adverse events. Overall, 4 of 21 subjects (19%) experienced a total of 8 TEAEs, with 3 of 16 subjects (19%) following active treatment and 1 of 5 (20%) following placebo. Of the 8 TEAEs reported, only 1 was suspected by the trial site primary investigator (PI) to be possibly treatment-related. The majority of the events (5) were mild (grade 1) in severity and 3 were moderate (grade 2). TEAEs are presented in Table 2. The most common TEAE was headache, experienced by 2 active-treatment subjects (13%). One case of headache of moderate severity (grade 2) was deemed to be possibly treatment related by the PI and had a reported onset of ∼10 minutes after dosing, resolving 36 hours later. The other case of headache (grade 1) was deemed to be unrelated to treatment by the PI and was reported 17 days after dosing, resolving 24 hours later. Other TEAEs reported included back pain, pruritis, papular rash, and skin exfoliation in 1 placebo-treated subject, and vessel puncture site ecchymosis at the site of attempted IV insertion in 1 active treatment subject. There were no detectable antidrug antibody responses to AB002 in any subject at either 14 or 28 days after dosing. Two subjects had positive titers for antibodies to WT thrombin that were present before dosing and did not change after dosing. Overall, AB002 appeared to be safe and well-tolerated.
Individual predose aPTT values ranged from 25 to 32 seconds in all subjects. AB002 produced a temporal, dose-dependent prolongation of the aPTT that peaked at 15 to 30 minutes and returned to baseline (±10%) by 2 to 4 hours (Figure 5A). The highest aPTT prolongation by dose level was 1.1-fold (0.5 µg/kg), 1.1-fold (1 µg/kg), 1.2-fold (2 µg/kg), and 1.5-fold (4 µg/kg). AB002 did not measurably impact prothrombin time (Figure 5C), platelet count, or fibrinogen levels. D-dimer levels were evaluated by ELISA in a subset of samples (highest dose level only) and there was no indication of an effect by AB002 (all samples below 500 ng/mL and no apparent time-dependent change; supplemental Figure 5A). Similarly, prothrombin fragment 1.2 levels analyzed by ELISA in a subset of samples from the same subjects appeared unaffected by AB002 and were all within the normal range (3-8 ng/mL; supplemental Figure 5B).
![Coagulation parameters from all subjects from the AB002 phase 1 clinical study. Four cohorts were administered a single dose of AB002 (cohort 1, 0.5 µg/kg; cohort 2, 1.0 µg/kg; cohort 3, 2.0 µg/kg; cohort 4, 4.0 µg/kg [n = 4 each dose level]) or placebo (all placebo-dosed subjects are grouped together [n = 5]). Plasma samples were collected at specific intervals to observe the effect of drug administration on (A) aPTT, (B) APC-PCI complex formation, (C) prothrombin time (PT) and (D) protein C levels. For aPTT and PT, samples were analyzed at 0, 5, 15, 30 minutes, 1, 2, 24 hours, and 14 and 28 days. For APC-PCI, samples were analyzed at 0, 5, 15, 30 minutes, and 1, 2, 4, 24 hours, and, for protein C, samples were analyzed at 0, 1, 2, 4, and 24 hours. There were no measurable levels of APC-PCI complex in placebo-dosed subjects, thus data are not shown. Black dashed lines indicate laboratory reference ranges and red dashed lines indicate clinically significant ranges. All data are expressed as mean plus or minus SEM.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/9/10.1182_blood.2019002771/4/m_bloodbld2019002771f5.png?Expires=1768854340&Signature=RjPGhPAHvE8vQzBKMnd9us1KrJbYNWYbcj-e5AulrZd2LbAE5iUcmk3xC~ErELTwbPtB0t9wZjvW8OWdc1sSyVoppBAQLKWAWdEY9k~uOZYSmFmxsqLITQMrStkogoY9SvSg3bBgV9xmMvLHaVQpw7rQwMdlpnzjwR8cLbXdf174fU16zmCoEqSRYS-SOWbaku8m0ipiulPWW4rANb3u1DLuy~DsIteFwBlubmh6x7u3wScNnoG197h1uXR4rdlC68gPDcOKOayC6a7Bjdtv6cCSmFRYaej5g6jWKzoDlyR~6oAGaWLhBsGoamJxnplHQyy1cXUJ~6pjkBFqhXPVLw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Coagulation parameters from all subjects from the AB002 phase 1 clinical study. Four cohorts were administered a single dose of AB002 (cohort 1, 0.5 µg/kg; cohort 2, 1.0 µg/kg; cohort 3, 2.0 µg/kg; cohort 4, 4.0 µg/kg [n = 4 each dose level]) or placebo (all placebo-dosed subjects are grouped together [n = 5]). Plasma samples were collected at specific intervals to observe the effect of drug administration on (A) aPTT, (B) APC-PCI complex formation, (C) prothrombin time (PT) and (D) protein C levels. For aPTT and PT, samples were analyzed at 0, 5, 15, 30 minutes, 1, 2, 24 hours, and 14 and 28 days. For APC-PCI, samples were analyzed at 0, 5, 15, 30 minutes, and 1, 2, 4, 24 hours, and, for protein C, samples were analyzed at 0, 1, 2, 4, and 24 hours. There were no measurable levels of APC-PCI complex in placebo-dosed subjects, thus data are not shown. Black dashed lines indicate laboratory reference ranges and red dashed lines indicate clinically significant ranges. All data are expressed as mean plus or minus SEM.
Coagulation parameters from all subjects from the AB002 phase 1 clinical study. Four cohorts were administered a single dose of AB002 (cohort 1, 0.5 µg/kg; cohort 2, 1.0 µg/kg; cohort 3, 2.0 µg/kg; cohort 4, 4.0 µg/kg [n = 4 each dose level]) or placebo (all placebo-dosed subjects are grouped together [n = 5]). Plasma samples were collected at specific intervals to observe the effect of drug administration on (A) aPTT, (B) APC-PCI complex formation, (C) prothrombin time (PT) and (D) protein C levels. For aPTT and PT, samples were analyzed at 0, 5, 15, 30 minutes, 1, 2, 24 hours, and 14 and 28 days. For APC-PCI, samples were analyzed at 0, 5, 15, 30 minutes, and 1, 2, 4, 24 hours, and, for protein C, samples were analyzed at 0, 1, 2, 4, and 24 hours. There were no measurable levels of APC-PCI complex in placebo-dosed subjects, thus data are not shown. Black dashed lines indicate laboratory reference ranges and red dashed lines indicate clinically significant ranges. All data are expressed as mean plus or minus SEM.
Plasma APC-PCI concentration results are presented in Figure 5B. APC-PCI was detectable in all subjects at the first time of sampling (5 min) postinjection and remained detectable throughout the sampling interval in all subjects up to 2 hours in the 0.5 µg/kg dose level and up to 4 hours in all other dose levels. Peak mean APC-PCI concentrations were observed at 30 minutes for all dose levels. Ascending doses of AB002 increased APC-PCI concentrations in an approximately dose-proportional manner. AB002 was also associated with an apparent modest reduction of protein C levels between 1 and 4 hours (Figure 5D). By 24 hours, protein C returned to predose levels in all groups. PCI levels were evaluated by ELISA in a subset of samples (highest dose only) and there was no measurable time-dependent depletion of PCI, consistent with the relatively high levels of PCI present in plasma and comparably low levels of APC-PCI measured in the subjects (data not shown).
Discussion
The present findings demonstrate for the first time that the thrombin mutant AB002 (E-WE thrombin) can rapidly interrupt acute experimental vascular graft thrombus propagation and prevent thrombo-occlusion without obvious signs or symptoms of toxicity. In the baboon acute thrombosis model, the antithrombotic activity of low-dose AB002 (≥1.25 µg/kg) appeared to be greater than tPA (1 mg/kg) and APC (50 µg/kg), while being indistinguishable from high-dose IV enoxaparin (2 mg/kg).
We have previously shown that infused exogenous APC has a significant antithrombotic effect in baboons only at doses that prolong aPTT well above the normal range.1 AB002 exhibits potent antithrombotic activity even at doses that cause only a slight aPTT prolongation. We thus contemplate that part of AB002’s antithrombotic effect may be caused by localized surface associated APC generation, spatially distant from the vascular endothelium. Because platelets bind thrombin via GPIb, and activated platelets have been shown to express low surface density, functionally active TM,28 we investigated whether high-density activated platelets that characterize the surface of thrombi could also serve as an active surface for supporting APC generation, independent of the endothelial TM. Indeed, the results of our experiments using ex vivo–formed, activated platelet aggregates were in agreement with the previous study showing that stimulated platelets can support APC generation. In our experiments, TM-blocking antibodies significantly decreased APC production, indicating that AB002-TM complexes were critical for promoting APC generation on platelets. Independently, these data also suggest a possible role for apolipoprotein E receptor 2 (ApoER2), the only known low-density lipoprotein receptor on platelets, as an accessory protein in thrombin- or AB002-mediated APC generation. We have previously shown that protein C binds ApoER2,31 which may help bring protein C to proximity of TM-AB002 or GPIb-AB002 complexes.
TAFI activation plays a role in thrombus stabilization and its activation is mediated by the thrombin/thrombomodulin complex, which also activates protein C. Importantly, critical thrombin residues for activation of TAFI and protein C differ,32 thus various thrombin mutants have been shown to differentially activate these 2 substrates. Western blot and quantitative activity assays confirmed that, although WT thrombin rapidly activates TAFI in the presence of TM, AB002 complexed with TM is a poor TAFI activator, in contrast to the single mutant E229K (E217K using the chymotrypsinogen numbering system), which showed an increase in TAFI activation compared with WT thrombin.33 In fact, likely by acting as a decoy enzyme or competing for TM binding, equimolar concentrations of AB002 inhibited thrombin-mediated TAFI activation by 25%, whereas a 50% reduction was observed with a fivefold molar excess. We also tested whether this inhibitory effect translated to AB002 augmenting clot lysis in vitro. These data indicated that AB002 can promote fibrinolysis by limiting TAFI activation, which may also contribute to the AB002 antithrombotic/thrombolytic activity observed in our primate model. We do acknowledge that without limiting the influence of APC in vivo, the extent to which TAFI inhibition or other mechanisms may contribute to AB002’s antithrombotic activity remains speculative.
Finally, AB002’s novel mechanism of action was found to be safe and well-tolerated in a phase 1 clinical trial conducted in healthy adult subjects. No serious adverse events occurred at any of the escalating doses from 0.5 µg/kg to 4 µg/kg, with only 1 potentially drug-related TEAE (headache) being observed. AB002 produced a modest dose-dependent aPTT prolongation with the highest values (<1.5-fold prolongation) measured at 15 to 30 minutes, returning to baseline by 2 to 4 hours. The transient increase in aPTT correlated with plasma APC-PCI levels. Prothrombin time was not affected by AB002, and no treatment-emergent cross-reacting antibodies to AB002 or thrombin were found in any subject.
Currently available anticoagulant and thrombolytic agents universally increase the risk or severity of bleeding, limiting their clinical utility. Modern guidelines restrict the use of systemically administered thrombolytics to patients with massive pulmonary embolism,34 patients with acute ST-elevation myocardial infarction unable to receive timely intervention,35,36 and acute ischemic stroke detected within several hours of onset,37 assuming a low bleeding risk in each case. Although unmet clinical needs persist in larger populations, attempts to expand the role of modern thrombolytic therapy have resulted in significant rates of hemorrhage that negate any clinical benefit. This is evidenced by attempts to evaluate the role of thrombolytic therapy in intermediate-risk pulmonary embolism where tPA was effective at preventing hemodynamic decompensation but significantly increased the risk of major bleeding (11.5%) including intracranial hemorrhage (2.0%).38 Likewise, modern forms of IV anticoagulation used to treat acute thrombotic disorders or to prevent device-associated thrombosis also significantly increase the risk of bleeding, while having subpar efficacy in certain populations.39 The unique antithrombotic activity of AB002 suggests it may be efficacious in many indications where there is a strong clinical need for safer and more effective therapies.
In conclusion, this study identifies new putative mechanisms by which low doses of AB002 can rapidly interrupt developing arterial-type thrombi. Given the proposed mechanisms of action, in which AB002’s effects appear to be primarily exerted locally at the site of propagating thrombi under shear flow, it may also spare the excessive systemic activities that lead to bleeding with traditional antithrombotic agents. Further clinical evaluations are in progress to determine the efficacy and safety of AB002 in patients.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors gratefully acknowledge Jennifer Johnson for providing technical assistance with the primates. HAPC 1555 antibody was generously provided by C. T. Esmon.
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grants HL095315 (E.I.T., A.G.), HL117589 (E.I.T., N.G.V.), HL049413, HL139554, and HL147821 (E.D.C.), and grant P51OD011092 from the National Institutes of Health Office of the Director (Oregon National Primate Research Center).
Authorship
Contribution: E.I.T., B.D.M., N.G.V., and A.G. designed the study, interpreted the data, and wrote the manuscript; M.W., B.D.M., L.A.P., and D.C.W. executed the experiments; and E.D.C., C.U.L., J.J.S., M.T.H., and O.J.T.M. critically reviewed the data and helped write the manuscript.
Conflict-of-interest disclosure: E.I.T., A.G., B.D.M., M.W., C.U.L., and N.G.V. are employees of Aronora, Inc. J.J.S. reports receiving consulting fees from Aronora Inc. A.G., O.J.T.M., N.G.V., E.I.T., and Oregon Health & Science University (OHSU) have a financial interest in Aronora, Inc. This potential conflict of interest has been reviewed and managed by the OHSU Conflict of Interest in Research Committee. The remaining authors declare no competing financial interests.
Correspondence: Erik I. Tucker, Aronora, Inc, 4640 SW Macadam Ave, Suite 200A, Portland, OR 97239; e-mail: erik.tucker@aronorabio.com.