

In this issue of Blood, report an intestinal organoid-based platform that re-creates genetic susceptibility to T-cell–mediated tissue injury as observed in a mouse model of intestinal graft-versus-host disease (GVHD), providing a roadmap for precision medicine.1
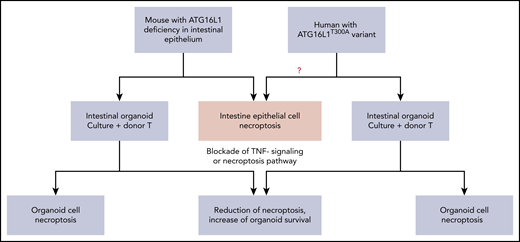
Organoid cultures as a bridge between mouse models and humans: ATG16L1 deficiency in both mouse and human intestinal epithelial cells augments the susceptibility toward necroptosis induced by alloreactive T cells or inflammatory cytokines.
Organoid cultures as a bridge between mouse models and humans: ATG16L1 deficiency in both mouse and human intestinal epithelial cells augments the susceptibility toward necroptosis induced by alloreactive T cells or inflammatory cytokines.
GVHD is a severe side effect of allogeneic hematopoietic cell transplantation (allo-HCT). GVHD of the gastrointestinal track (gut-GVHD) has an adverse impact on the outcome of allo-HCT.2 Alloreactive donor T cells initiate GVHD, including gut-GVHD. Previous reports have focused on the pathogenicity of donor alloreactive T cells; that is, how donor immune cells launch inflammatory attacks on the host tissues.3-5 However, Matsuzawa-Ishimoto and colleagues focus on the other side of the coin, that is, genetic susceptibility of host tissues to respond to the immune attacks. This report provides insights on how host-tissue genetic susceptibility is involved in regulating GVHD severity and how ex vivo organoid culture can serve as a platform for developing personalized therapy.
First, mice with ATG16L1 deficiency in intestinal epithelium can serve as a model of host-parenchymal tissue genetic susceptibility regulating GVHD severity. Autophagy gene ATG16L1 prevents necroptosis in the intestinal epithelium.6 Necroptosis is a programmed necrosis mediated by receptor interacting protein kinase-3 (RIPK3) and RIPK1 as well as their substrate mixed lineage kinase-like.7 Different from programmed cell death of apoptosis, necroptosis triggers tissue inflammation.7 Matsuzawa-Ishimoto et al showed that mouse recipients with ATG16L1 deficiency in the intestinal epithelial cells had increased GVHD severity and mortality.
These recipients had loss of Paneth cells in the small intestine, without skewing donor T-cell differentiation.1 RIPK3 deficiency in the ATG16L1-deficient recipient or administration of RIPK1 inhibitor GSK547 reduced GVHD severity and increased intestinal Paneth cell and epithelial cell survival.1 Ex vivo intestinal organoid culture with alloreactive donor T cells induced necroptosis of intestinal organoid cells from the ATG16L1-deficient mice via interferon-γ (IFN-γ) and/or tumor necrosis factor-α (TNF-α).1 These observations indicate that inflammatory cytokines from alloreactive T cells can trigger necroptosis in recipients with a defect in protective genes such as ATG16L1. These studies also emphasize the involvement of host tissue in regulating GVHD, opening a new area of research to evaluate other protective genes in the parenchymal cells of GVHD target tissues.
Second, host-tissue genetic susceptibility differences may help explain some of the discrepancies in disease severity and therapy efficacy among animal models and humans. Acute GVHD, especially acute gut-GVHD, was thought to be mediated by T helper 1 (Th1) and Tc1 cells and their production of IFN-γ and TNF-α in mouse models of allo-HCT.3 However, after in vivo depletion of CD4+ T cells by anti-CD4 monoclonal antibody, higher levels of serum IFN-γ and TNF-α did not cause gut-GVHD in wild-type (WT) recipients.4 Interestingly, Matsuzawa-Ishimoto et al showed that although donor Th1 and Tc1 cells induced moderate GVHD lethality in WT recipients, the same donor Th1 and Tc1 cells induced severe lethality in recipients with ATG16L1 deficiency in intestinal epithelial cells. These observations indicate that GVHD severity is determined not only by the strength of inflammatory attack from donor T cells or inflammatory cytokines but also by genetic factors in the target tissues, such as WT recipients vs ATG16L-deficient recipients.
It would be of interest to find out whether in vivo depletion of donor CD4+ T cells prevents acute gut-GVHD in the ATG16L1-deficient mouse recipients. If there is no prevention, it would be consistent with Matsuzawa-Ishimoto and colleagues’ observation that mouse recipients with intestinal ATG16L1 deficiency had enhanced gut-GVHD mediated by IFN-γ and TNF-α from Th1/Tc1 cells. Similarly, neutralizing IFN-γ or TNF-α was effective in ameliorating gut-GVHD in some patients but not others.8,9 It would also be of interest to find out whether the different efficacies are associated with the different ATG16L1 variants of the patients.
Interestingly, Matsuzawa-Ishimoto et al showed that intestinal organoids from patients with ATG16L1T300A/T300A risk alleles had markedly increased necroptosis when cocultured with TNF-α or donor T cells.
Third, ex vivo organoid culture can link mouse models to human pathogenesis and provide a platform for precision medicine. As depicted in the diagram (see figure), mouse recipients with ATG16L1 deficiency in intestinal epithelium had augmented gut-GVHD with enhanced intestinal epithelial necroptosis after allo-HCT, which could be recapitulated in an ex vivo organoid culture system.1 The death of the cultured organoid mediated by necroptosis was associated with an IFN signature, as measured with RNA-Seq analysis.1 Blockade of the IFN signaling pathway by the JAK2 inhibitor Ruxolitinib or blockade of necroptosis pathway by RIPK1 inhibitor GSK547 markedly reduced necroptosis and increased survival of cultured intestinal organoids derived from ATG16L1-deficient mice.1
Matsuzawa-Ishimoto et al were also able to set up intestinal organoid cultures from fresh as well as frozen human intestinal tissues. Coculture with donor T cells or TNF-α induced death of the cultured human organoids. Remarkably, organoid cell death was markedly increased when intestinal tissues were from patients with ATG16L1T300A/T300A risk alleles, and blockade of IFN signaling or necroptosis greatly increased the organoid cell survival1 (see figure). Therefore, the GVHD protective role of intestinal epithelial cell expression of ATG16L1 in mouse and human can be both demonstrated by ex vivo organoid cultures. More importantly, the organoid cultures allow for screening new drugs with both mouse and human tissues for targeting the same inflammatory pathways. This approach allows for linked observations from ex vivo assays to in vivo mouse models or vice versa.
Finally, GVHD and other inflammatory diseases have tissue-specific pathogenesis.3 Ex vivo organoid culture systems can use different tissues, including intestine, liver, lung, heart, and skin.10 Therefore, ex vivo organoid culture systems can serve as a platform for testing pathogenic or protective lymphocytes and drug candidates. The combination of mouse models and ex vivo organoid culture systems may ultimately serve as a platform for personalized medicine.
Conflict-of-interest disclosure: The author declares no competing financial interests.

