

Key Points
X-linked FANCB pathogenic variants predominantly cause early-onset bone marrow failure and severe congenital abnormalities.
Biochemical and cell-based assays of causative variants reveal functional properties of FANCB that are associated with clinical severity.

Abstract
Fanconi anemia (FA) is the most common genetic cause of bone marrow failure and is caused by inherited pathogenic variants in any of 22 genes. Of these, only FANCB is X-linked. We describe a cohort of 19 children with FANCB variants, from 16 families of the International Fanconi Anemia Registry. Those with FANCB deletion or truncation demonstrate earlier-than-average onset of bone marrow failure and more severe congenital abnormalities compared with a large series of FA individuals in published reports. This reflects the indispensable role of FANCB protein in the enzymatic activation of FANCD2 monoubiquitination, an essential step in the repair of DNA interstrand crosslinks. For FANCB missense variants, more variable severity is associated with the extent of residual FANCD2 monoubiquitination activity. We used transcript analysis, genetic complementation, and biochemical reconstitution of FANCD2 monoubiquitination to determine the pathogenicity of each variant. Aberrant splicing and transcript destabilization were associated with 2 missense variants. Individuals carrying missense variants with drastically reduced FANCD2 monoubiquitination in biochemical and/or cell-based assays tended to show earlier onset of hematologic disease and shorter survival. Conversely, variants with near-normal FANCD2 monoubiquitination were associated with more favorable outcome. Our study reveals a genotype-phenotype correlation within the FA-B complementation group of FA, where severity is associated with level of residual FANCD2 monoubiquitination.
Introduction
Fanconi anemia (FA) is an inherited disorder caused by DNA repair deficiency that results in progressive bone marrow failure, leading to aplastic anemia. FA is also associated with congenital abnormalities and a highly elevated risk of acute myeloid leukemia and head and neck cancers.1 In a large series of patients, the average age at onset of hematologic abnormalities was reported to be 7.6 years,2 and approximately two-thirds of patients exhibited ≥1 congenital abnormalities.3 At least 22 FA-causing genes have been identified, termed FANCA-FANCW,4 but only FANCB (MIM# 300515) is X-linked recessive.5 This makes it especially important to identify pathogenic FANCB variants and classify them as inherited or de novo to provide proper genetic counseling. Variants in FANCB account for ∼4% of all male cases of FA6 and are often associated with severe congenital abnormalities resembling vertebral abnormalities, anal atresia, cardiac defects, tracheoesophageal fistula, esophageal atresia, renal abnormalities, or limb abnormalities, along with hydrocephalus (VACTERL-H).7,8 The course of hematologic disease and severity of VACTERL-H or any genotype-phenotype correlations among FANCB individuals remain to be elucidated.
FANCB protein is a component of the FA core complex that also contains the products of 6 other FANC genes (A, C, E, F, G, and L), together with FA-associated proteins (FAAP100 and FAAP20).9 The FA core complex is necessary to signal the existence of stalled replication forks, caused by DNA interstrand crosslinks, covalent bonds between Watson and Crick DNA strands. It is required for monoubiquitination of the DNA binding proteins FANCD2 and FANCI, which form the ID2 heterodimer.10 In particular, FANCB, FANCL, and FAAP100 are the critical minimal components required for in vitro monoubiquitination of ID2.11,12 Whereas FANCL is the RING E3 ligase necessary for ubiquitination to occur, FANCB and FAAP100 play an enigmatic role in the reaction. FANCT/UBE2T is the cellular E2 ligase necessary for ID2 ubiquitination.13
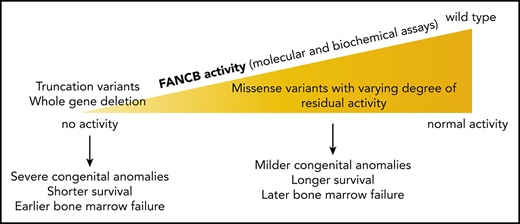
Very few studies have explored the function of the FANCB protein, despite its identification as a critical enzymatic component of the FA core complex. This is in part because the protein contains no known predicted domains and also because it is intractable to work with in isolation. It has been shown that coexpression and purification of a recombinant complex of FANCB, FANCL, and FAAP100 (BL100 complex) can overcome these difficulties,11,12,14 and this reflects a similar interdependence of stability for these 3 proteins in human cells.5,15,16 Recombinant BL100 complex is twice its predicted mass, reflective of a dimerization of the complex through the FANCB subunit.12 BL100 can also interact independently with the CEF (FANCC, FANCE, and FANCF) and AG20 (FANCA, FANCG, and FAAP20) subcomplexes of the FA core complex. Here, through biochemical and cell-based assays, we demonstrate the central role of FANCB in FA core complex assembly and activity and establish how variants identified in affected individuals disrupt DNA damage signaling in FA. We present molecular diagnosis, functional evaluation of the variants, and clinical presentations of the largest cohort of individuals with FANCB variants yet described. We find that individuals with missense variants, in general, had a delayed onset of hematologic disease and longer overall survival than those with whole gene deletion (WGD) or truncation variants, which correlate with the extent of residual function retained by the variant FANCB protein.
Methods
Study participants
The study participants included individuals diagnosed with FA and their family members enrolled in the International Fanconi Anemia Registry (IFAR), after written informed consent/assent. The Institutional Review Board of the Rockefeller University (New York, NY) approved these studies. The Office of Human Subjects Research at the National Institutes of Health and Institutional Review Board of the National Human Genome Research Institute approved the reception of deidentified cell lines and DNA samples from The Rockefeller University and analysis of the underlying molecular variants.
Identification of disease-causing FANCB variants
Genomic DNA was isolated from peripheral blood, fibroblasts, or Epstein-Barr virus–immortalized lymphoblastoid cell lines (LCLs). Targeted next-generation sequencing, Sanger sequencing, array comparative genomic hybridization (aCGH), and single-nucleotide polymorphism arrays were employed to identify sequence variants as described earlier17 and are further outlined in supplemental methods.
Reverse transcription PCR
Total RNA from cell lines derived from affected individuals was obtained using RNeasy Plus Mini Kit (Qiagen). Complementary DNA synthesis was performed using SuperScript III or IV First-Strand Synthesis System (Thermo Fisher Scientific). Screening for aberrant FANCB splice products was performed, as described further in supplemental methods.
Quantitative fluorescence PCR
A modified version of the previously described quantitative fluorescence polymerase chain reaction (PCR) method was used to measure the relative levels of aberrant splice or missense product vs normal splice product.18,19 A FAM-labeled primer (M13F-FAM) was included to generate fluorescently labeled products. Primers to generate splice products with similar sizes were designed and are listed in supplemental Table 1. Details of PCR reactions are provided in supplemental methods.
Cell culture
Fibroblasts were cultured in Dulbecco’s modified Eagle medium (Gibco) plus 15% fetal bovine serum (Atlanta Biologicals), 1% Pen Strep (Gibco), 1% GlutaMAX (Gibco), and 1% MEM nonessential amino acids (Gibco). LCLs were cultured in RPMI 1640 (Gibco) plus 20% fetal bovine serum, 1% Pen Strep, and 1% GlutaMAX. Fibroblast cell lines were transformed and/or immortalized by expression of HPV16 E6E7 and the catalytic subunit of human telomerase, respectively.
Complementation of FANCB-deficient cell lines with WT or variant FANCB
Cloning, lentiviral vector preparation, and complementation experiments of the wild-type (WT) FANCB and missense variants were performed as described earlier20 and are further outlined in supplemental methods.
Recombinant FANCB protein copurification
FANCB variant was cloned into pFL-FANCB-FANCL-FAAP100, and purified protein was generated from resultant baculovirus, as described by Swuec et al14 and summarized in the supplemental methods.
In vitro ubiquitination experiments
All protein purifications and reactions were performed exactly as described by van Twest et al12 and summarized in supplemental methods, substituting variant FANCB-containing BL100 complexes where appropriate. StrepII-FANCD2 and Flag-FANCI were detected with αStrepII tag (ab76949; Abcam) or αFlag-tag (OAEA00002; Aviva Systems Biology) antibodies.
Statistical analyses of clinical data
The statistical analyses of clinical data for time to death and time to hematologic disease were performed as outlined in supplemental methods.
Results
Wide spectrum of inherited or de novo FANCB pathogenic variants cause FA
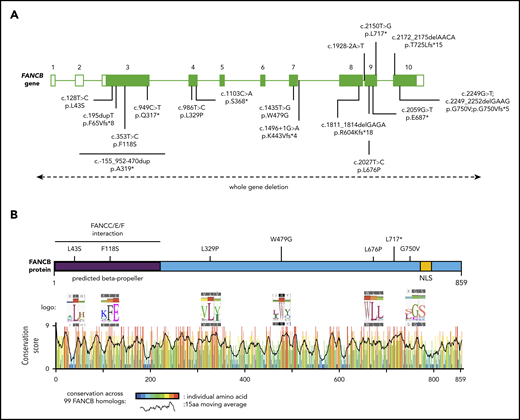
In a comprehensive effort toward molecular diagnosis of FA, we identified disease-causing FANCB variants in 19 individuals from 16 families enrolled in the IFAR, which accounts for 4.0% of genotyped males with FA (Figure 1A; supplemental Figure 1; Table 1). Table 1 describes the clinical information and genotypes of these 19 individuals from the IFAR and 2 additional individuals (individuals 9 and 21) reported on previously by others.8,21 Among the 19 IFAR individuals, we observed 2 large deletions, 590 kb (individuals 1 and 2) and 520 kb (individual 3), encompassing the FANCB gene in 2 families. Single-nucleotide polymorphism arrays and aCGH showed that these deletions included the entire FANCB gene as well as an adjacent upstream gene, MOSPD2, and a downstream gene, GLRA2 (supplemental Figure 2). Furthermore, we identified sequence variants predicted to generate a truncated protein in 9 individuals, including 2 variants affecting splice junctions (individuals 4 and 5), 3 resulting in a nonsense codon (individuals 6-8), and 4 indels from 3 families (individuals 11-14). Individual 15 carried 2 overlapping de novo variants, a missense (NM_001018113:c.2249G>T; p.G750V) and an indel (NM_001018113:c.2249_2252delGAAG p.G750Vfs*5). The proportion of the missense variant was observed to decrease from 80% in blood to 53% in the LCL, suggesting positive growth selection for cells with the indel variant (supplemental Table 2). Five individuals from 4 families carried missense variants (individuals 16-20). All missense variants appeared in highly conserved residues across 99 FANCB orthologs (Figure 1B; supplemental Table 3). Additionally, we previously reported individual 10,20 who carried an unstable 9154-bp intragenic duplication, exhibiting mosaicism in both the proband and the carrier mother; this individual is not discussed further in this report. It is notable that each of the families in this study had a unique pathogenic variant, with no evidence of a common founder.
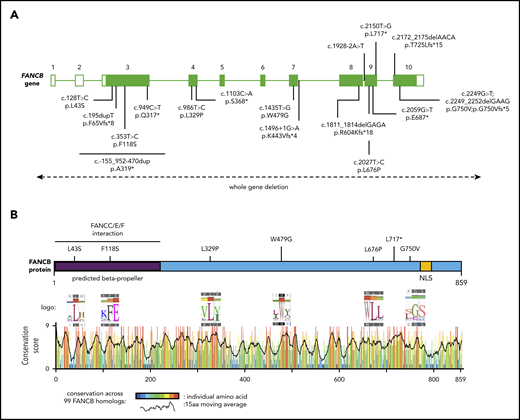
Schematic of FANCB gene and protein with identified variants. (A) Schematic of FANCB gene with intron-exon organization, complementary DNA coordinates, and the predicted amino acid changes for each identified variant. One IFAR family with a duplication c.-155_952-470dup (p.A319*) has been reported earlier,20 and the duplicated region is shown as a solid line. Two variants from published reports, F118S21 and L717*,8 are indicated, because their functional status was evaluated in this study. The WGDs extending beyond FANCB are indicated by a dotted line with arrows at the end. For the splice junction variant in intron 7, c.1496+1G>A, the predicted protein change is based on RNA analysis, whereas lack of RNA prevented identifying the consequences for the protein of the intron 8 variant, c.1928-2A>T. (B) Schematic of FANCB protein indicating location of missense variants. The predicted structural motifs, β-propeller, and nuclear localization signal (NLS) are shown. The conservation score plot of FANCB across 99 vertebrate homologs is shown below. For each amino acid that is altered in an individual with FA, the sequence logo derived from 99 FANCB vertebrate homologs is shown for the affected and the adjacent amino acids. The consensus sequence and the human WT FANCB sequence are above and below the logo, respectively. The location of the nonsense variant p.L717* is also shown.
Schematic of FANCB gene and protein with identified variants. (A) Schematic of FANCB gene with intron-exon organization, complementary DNA coordinates, and the predicted amino acid changes for each identified variant. One IFAR family with a duplication c.-155_952-470dup (p.A319*) has been reported earlier,20 and the duplicated region is shown as a solid line. Two variants from published reports, F118S21 and L717*,8 are indicated, because their functional status was evaluated in this study. The WGDs extending beyond FANCB are indicated by a dotted line with arrows at the end. For the splice junction variant in intron 7, c.1496+1G>A, the predicted protein change is based on RNA analysis, whereas lack of RNA prevented identifying the consequences for the protein of the intron 8 variant, c.1928-2A>T. (B) Schematic of FANCB protein indicating location of missense variants. The predicted structural motifs, β-propeller, and nuclear localization signal (NLS) are shown. The conservation score plot of FANCB across 99 vertebrate homologs is shown below. For each amino acid that is altered in an individual with FA, the sequence logo derived from 99 FANCB vertebrate homologs is shown for the affected and the adjacent amino acids. The consensus sequence and the human WT FANCB sequence are above and below the logo, respectively. The location of the nonsense variant p.L717* is also shown.
Availability of parental DNA for 13 of 16 IFAR families allowed us to infer the origin of the pathogenic variant. Nine were inherited (12 individuals), whereas 4 were presumed to be de novo (ie, arose during gametogenesis in a parent or very early in embryogenesis; Table 1). We performed deep sequencing of the variant region in the proband and parents using the MiSeq method (supplemental Table 4). At a read depth in the range of 223 000 to 609 000, we confirmed the variants to be absent in the parents and de novo in the probands (supplemental Table 2). Despite being de novo in origin, blood mosaicism was not observed, and the variant frequency was ∼100% in 3 individuals. In the fourth individual (individual 15), there were 2 overlapping de novo variants. Also, through deep sequencing, we confirmed that mosaicism was not present in any of the 5 tested probands with inherited variants or in their parents/siblings (supplemental Table 5).
FANCB RNA analysis reveals aberrant splice and unstable transcripts
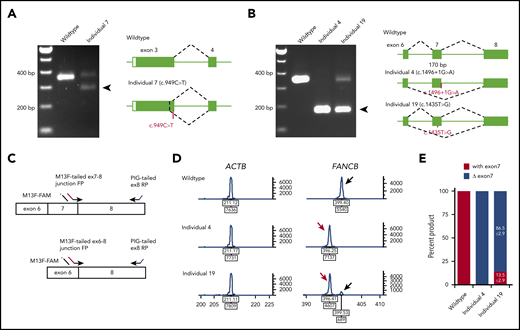
Availability of cell lines for all individuals except individual 5 (c.1928-2A>T) allowed us to analyze the splicing and stability of FANCB transcripts. Aberrant splicing products were observed in individuals 4, 7, and 19. Individual 7, carrying a nonsense variant (c.949C>T; p.Q317*) near the end of exon 3, generated a second transcript that eliminates 74 bp from the end of exon 3 (p.G294Cfs*3; Figure 2A). Individual 4, carrying a canonical splice variant (c.1496+1G>A), generated a frameshift transcript as a result of skipping of exon 7 (170 bp; p.K443Vfs*4; Figure 2B). Individual 19, carrying a missense variant in exon 7 (c.1435T>G; p.W479G), generated an additional transcript without exon 7 (Figure 2B). The relative levels of transcripts with and without exon 7 were 13.5% and 86.5%, respectively (Figure 2C-E).
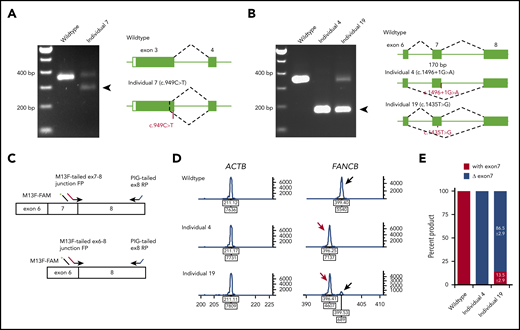
Aberrant FANCB splice products and their quantitation. (A) Reverse transcription PCR (RT-PCR) analysis of fibroblast cell line RNA from individual 7 with c.949C>T (p.Q317*) variant in exon 3. The gel shows, along with the predicted size product, an additional smaller product (arrow) that lacked the last 74 bp from the end of exon 3 (c.878_951del74; p.G294Cfs3*). Both products encode truncated proteins. RNA from a fibroblast cell line from an unaffected individual was used as WT control. (B) RT-PCR analysis of fibroblast cell line RNA from individuals 4 and 19 carrying splice junction c.1496+1G>A and missense c.1435T>G (p.W479G) variants, respectively. Individual 4 RNA shows exclusively exon 7 skipping (c.1327_1496del170; p.K443Vfs*4) (arrow). Individual 19 also shows exon 7 skipping along with a normal size product. (C) Schematics of quantitative fluorescence RT-PCR (qf-RT-PCR) for relative quantification of transcripts with and without exon 7. Two FANCB-specific forward primers (FPs) that bind to either exon 7-8 junction or exon 6-8 junction and a reverse primer (RP) that binds to exon 8 were designed to amplify products from transcripts with (top) or without exon 7 (bottom), respectively. The product with exon 7 is 3 bp longer than that from the product without exon 7. FAM-labeled M13 FP (M13F-FAM) was included to generate fluorescently labeled products. (D) Representative qf-RT-PCR profiles from individuals 4 and 19. Product size and intensity are on the x- and y-axes, respectively; both numbers are shown under each peak. The left panel shows products from ACTB, an internal transcript control. The right panel shows FANCB products. The fragment size for products with exon 7 (black arrow) and without exon 7 (red arrow) appeared around 399 and 396 bp, respectively. Data from WT, individual 4, and individual 19 are presented on the top, middle, and bottom rows, respectively. (E) Percentage of transcripts with or without exon 7; average from 8 independent assays.
Aberrant FANCB splice products and their quantitation. (A) Reverse transcription PCR (RT-PCR) analysis of fibroblast cell line RNA from individual 7 with c.949C>T (p.Q317*) variant in exon 3. The gel shows, along with the predicted size product, an additional smaller product (arrow) that lacked the last 74 bp from the end of exon 3 (c.878_951del74; p.G294Cfs3*). Both products encode truncated proteins. RNA from a fibroblast cell line from an unaffected individual was used as WT control. (B) RT-PCR analysis of fibroblast cell line RNA from individuals 4 and 19 carrying splice junction c.1496+1G>A and missense c.1435T>G (p.W479G) variants, respectively. Individual 4 RNA shows exclusively exon 7 skipping (c.1327_1496del170; p.K443Vfs*4) (arrow). Individual 19 also shows exon 7 skipping along with a normal size product. (C) Schematics of quantitative fluorescence RT-PCR (qf-RT-PCR) for relative quantification of transcripts with and without exon 7. Two FANCB-specific forward primers (FPs) that bind to either exon 7-8 junction or exon 6-8 junction and a reverse primer (RP) that binds to exon 8 were designed to amplify products from transcripts with (top) or without exon 7 (bottom), respectively. The product with exon 7 is 3 bp longer than that from the product without exon 7. FAM-labeled M13 FP (M13F-FAM) was included to generate fluorescently labeled products. (D) Representative qf-RT-PCR profiles from individuals 4 and 19. Product size and intensity are on the x- and y-axes, respectively; both numbers are shown under each peak. The left panel shows products from ACTB, an internal transcript control. The right panel shows FANCB products. The fragment size for products with exon 7 (black arrow) and without exon 7 (red arrow) appeared around 399 and 396 bp, respectively. Data from WT, individual 4, and individual 19 are presented on the top, middle, and bottom rows, respectively. (E) Percentage of transcripts with or without exon 7; average from 8 independent assays.
The quantitative analysis of the LCL from individual 15 revealed the presence of indel variant transcripts only, whereas genomic DNA analysis showed the presence of both the indel and missense variants (supplemental Figure 3A-B), suggesting that the missense variant transcript is highly unstable. The unstable nature of this variant may be the reason behind the observed positive growth selection for cells with the indel variant from blood to LCL (supplemental Table 2).
Functional evaluation of constructs expressing missense variants in a FANCB-null cell line
Unlike the nonfunctional nature of a truncated protein variant, the effect of a missense variant is difficult to determine without functional evaluation. We assessed the function of all the identified FANCB missense variants using cell lines derived from the affected individuals and overexpression of the variant in an FANCB-null cell line. Cell lines from all individual with missense variants lacked FANCD2 ubiquitination upon mitomycin C treatment, confirming the absence of a functional FA core complex (supplemental Figure 4A-B). FANCD2 ubiquitination was completely restored upon complementation with WT FANCB expression (supplemental Figure 4A), identifying the FANCB variants as causative for FA phenotypes in these individuals. The cell line from individual 15 (RA2951), who carried both a missense and an indel variant, showed a low level of FANCD2 ubiquitination.
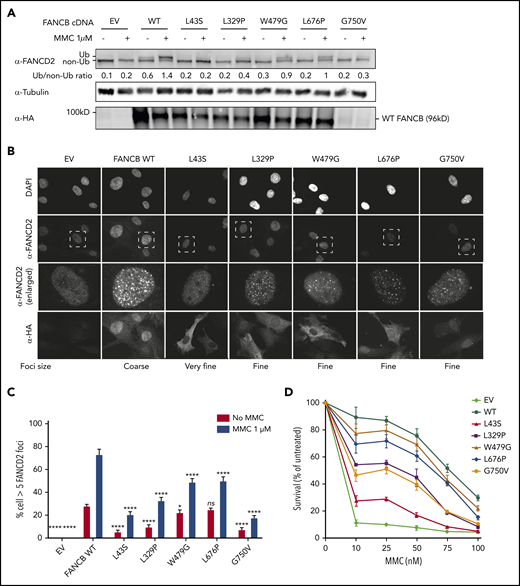
We next introduced complementary DNA expression constructs into an FANCB-null cell line and evaluated FANCB function using 3 assays posttreatment with mitomycin C: FANCD2 ubiquitination, FANCD2 foci formation, and cell viability (Figure 3). FANCD2 ubiquitination was nearly absent in the cell lines expressing the L43S and L717* variants, minimal in the L329P, G750V, and F118S cell lines, and comparable to WT in the W479G and L676P cell lines (Figure 3A; supplemental Figure 5B). The FANCD2 foci formation assay (Figure 3B-C; supplemental Figure 5C) and cell survival assay (Figure 3D; supplemental Figure 5A) also revealed variable activity of the variants: L43S and L717* showed very little function, L329P and F118S had moderate activity, and W479G and L676P displayed substantial activity. The high activity of W479G and L676P was somewhat surprising, but we conclude that the high overexpression achieved with lentiviral expression is able to support high function, although not complete restoration, of the repair pathway. The G750V-complemented cell line showed little function in these assays; however, we found that the lentiviral transcript carrying the c.2249G>T (G750V) variant was expressed at a 3.5-fold lower level than the WT FANCB transcript (supplemental Figure 3C). This resulted in reduced expression of G750V protein, indicating that the c.2249G>T variant may destabilize the transcript. Except for G750V, which shows globally low expression of HA-FANCB, the remaining missense variants also had reduced nuclear localization (supplemental Figure 6).
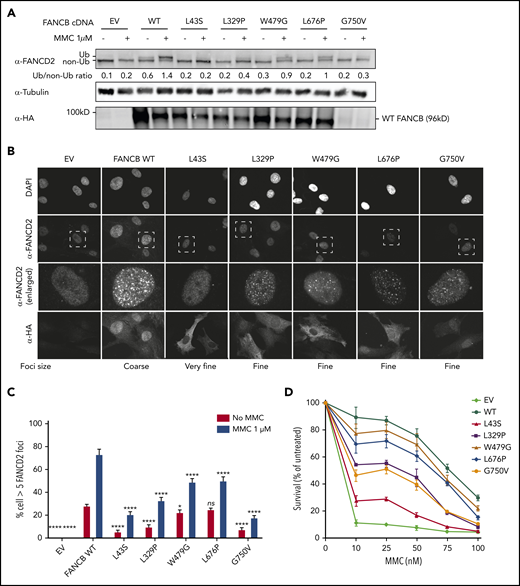
Overexpression of missense variants in FANCB-null fibroblasts show varying degree of residual function. hemagglutinin (HA)-tagged complementary DNA of FANCB WT or 5 missense variants were overexpressed by lentiviral vector in FANCB-null fibroblasts, followed by functional analysis. (A) Western blot of FANCD2, tubulin, and HA-FANCB. Near-normal FANCD2 ubiquitination was observed upon mitomycin C (MMC) exposure in cells expressing p.W479G and p.L676P, whereas cells expressing p.L43S showed almost no FANCD2 ubiquitination. Expression of p.L329P and p.G750V resulted in low level of FANCD2 ubiquitination. HA-FANCB p.G750V was unstable. Tubulin was used as loading control. Relative ratio of ubiquitinated FANCD2 band to nonubiquitinated FANCD band was measured for each variant. (B) Immunofluorescence of FANCD2 and HA-FANCB. Cells expressing HA-FANCB missense variants displayed smaller and fewer FANCD2 foci than cells expressing WT FANCD2 (original magnification ×630 oil immersion). (C) Quantification of FANCD2 foci–positive cells. One hundred cells were counted in triplicate per each experiment. Three independent experiments were performed. Statistical analysis was performed using one-way analysis of variance followed by Dunnett’s multiple comparison test. (D) Fibroblasts expressing FANCB WT or missense variants were treated with corresponding doses of MMC. Surviving cells were counted, and survival was calculated relative to untreated cells. Three independent experiments were performed. A graph from a representative experiment is shown. *P < .05, ****P < .001. DAPI, 4′,6-diamidino-2-phenylindole; EV, empty vector; ns, not significant.
Overexpression of missense variants in FANCB-null fibroblasts show varying degree of residual function. hemagglutinin (HA)-tagged complementary DNA of FANCB WT or 5 missense variants were overexpressed by lentiviral vector in FANCB-null fibroblasts, followed by functional analysis. (A) Western blot of FANCD2, tubulin, and HA-FANCB. Near-normal FANCD2 ubiquitination was observed upon mitomycin C (MMC) exposure in cells expressing p.W479G and p.L676P, whereas cells expressing p.L43S showed almost no FANCD2 ubiquitination. Expression of p.L329P and p.G750V resulted in low level of FANCD2 ubiquitination. HA-FANCB p.G750V was unstable. Tubulin was used as loading control. Relative ratio of ubiquitinated FANCD2 band to nonubiquitinated FANCD band was measured for each variant. (B) Immunofluorescence of FANCD2 and HA-FANCB. Cells expressing HA-FANCB missense variants displayed smaller and fewer FANCD2 foci than cells expressing WT FANCD2 (original magnification ×630 oil immersion). (C) Quantification of FANCD2 foci–positive cells. One hundred cells were counted in triplicate per each experiment. Three independent experiments were performed. Statistical analysis was performed using one-way analysis of variance followed by Dunnett’s multiple comparison test. (D) Fibroblasts expressing FANCB WT or missense variants were treated with corresponding doses of MMC. Surviving cells were counted, and survival was calculated relative to untreated cells. Three independent experiments were performed. A graph from a representative experiment is shown. *P < .05, ****P < .001. DAPI, 4′,6-diamidino-2-phenylindole; EV, empty vector; ns, not significant.
Efficiency of missense variants in recombinant FA core complex assembly and in vitro FANCD2/FANCI ubiquitination
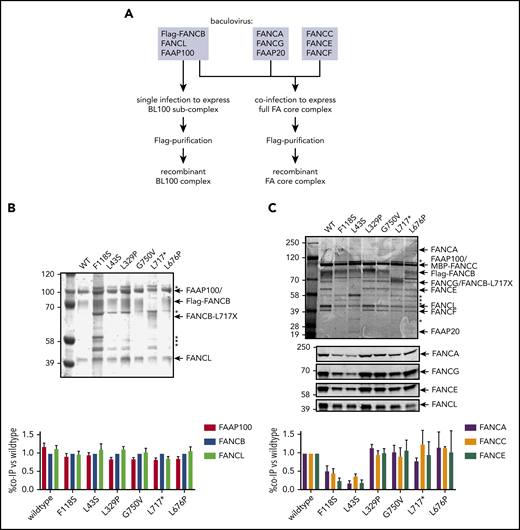
To define how FANCB variants directly affect the assembly of the FA core complex and subcomplexes, we used an in vitro reconstitution assay and biochemical test of FA core complex assembly and activity in ubiquitination of FANCD2 and FANCI proteins. In addition to the missense variants identified through the IFAR, we included F118S, the only other missense variant reported in the literature,21 and L717*, the truncation variant missing only the last 142 amino acids.8 The FA core complex is made up of 3 subcomplexes: BL100, AG20, and CEF.22 Baculoviral expression constructs of FA core complex proteins were used for coexpression of either BL100 subcomplex proteins or all the 9 proteins of the FA core complex (Figure 4A). Flag-tagged WT or missense variant FANCB protein was used as bait to purify protein complexes, resolved using PAGE, and the components were then quantitated. The assembly of the BL100 subcomplex was not affected by the missense variants or the 142 amino acid C-terminal truncation variant (L717*; Figure 4B). However, the L43S and F118S variants showed significantly reduced AG20 and CEF association (Figure 4C). Using FFAS-3D,23 this region of the protein was predicted to have structural homology to other proteins containing β-propeller structures, a known protein/protein interaction motif. SMURFLite analysis, which predicts β-propeller structures more stringently,24 concurred with high confidence (supplemental Figure 7). A structural model of the Gallus gallus FA core complex also assigns the N-terminus of chicken FANCB to a β-propeller structure, which was hypothesized to independently engage the CEF and AG20 modules.25 The remaining variants, which lie outside of this region, L329P, G750V, and L676P and the L717* truncation, retained WT levels of FA core complex assembly (Figure 4C).
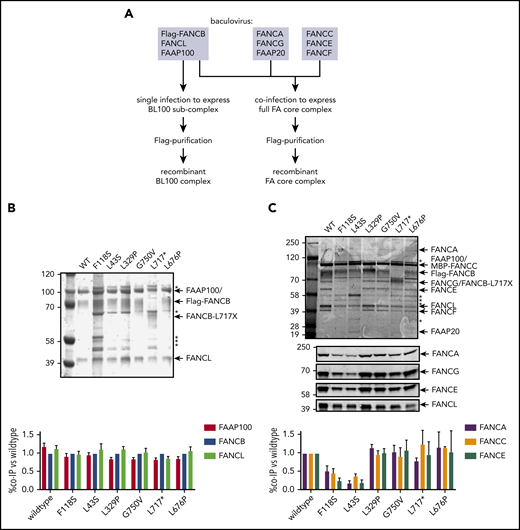
N-terminal FANCB variants affect FA core complex assembly. (A) Schematic for protein purifications of human FANCB complexes purified from baculovirus infected insect cells. (B) Coomassie blue–stained sodium dodecyl sulfate–polyacrylamide gel electrophoresis gel of affinity-purified FANCB-FANCL-FAAP100 (BL100) complex and bar graph (n = 2 experiments) indicating relative molar amount of FANCL and FAAP100 copurified with WT of variant Flag-FANCB protein as bait. (C) As in panel B but using full FA core complex. Western blots for FANCA, FANCG, FANCE, and FANCL are shown to identify individual Coomassie-stained bands. Asterisks represent contaminant proteins that bind to Flag-affinity resin.
N-terminal FANCB variants affect FA core complex assembly. (A) Schematic for protein purifications of human FANCB complexes purified from baculovirus infected insect cells. (B) Coomassie blue–stained sodium dodecyl sulfate–polyacrylamide gel electrophoresis gel of affinity-purified FANCB-FANCL-FAAP100 (BL100) complex and bar graph (n = 2 experiments) indicating relative molar amount of FANCL and FAAP100 copurified with WT of variant Flag-FANCB protein as bait. (C) As in panel B but using full FA core complex. Western blots for FANCA, FANCG, FANCE, and FANCL are shown to identify individual Coomassie-stained bands. Asterisks represent contaminant proteins that bind to Flag-affinity resin.
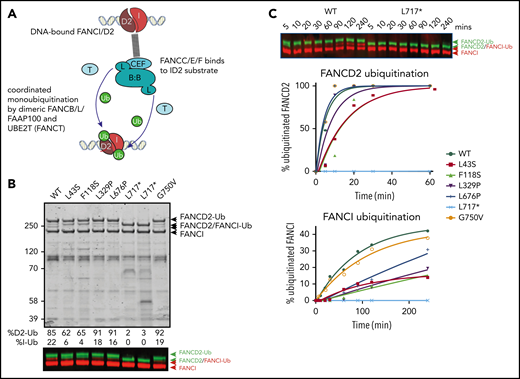
Because several variant BL100 complexes failed to properly interact with the remainder of the core complex, we evaluated their effect on in vitro ubiquitination of FANCD2 and FANCI using BL100 complexes purified in isolation and then added to other core complex components (Figure 5A). The levels of monoubiquitylation of FANCD2 and FANCI were measured using western blot analysis. As predicted by poor FA core complex assembly, the L43S and F118S variants showed reduced FANCD2 and FANCI monoubiquitination (Figure 5B). As previously observed, the kinetics of FANCI monoubiquitination were slower than those for FANCD2, and the N-terminal changes effected both reactions proportionally. The in vitro ubiquitination activity of complexes with L329P and L676P variants resembled WT FANCB for FANCD2 monoubiquitination; however, these had a slower rate of FANCI monoubiquitination (Figure 5C). Unexpectedly, given that L717* still forms an intact FA core complex, this truncation variant was completely defective in both FANCD2 and FANCI monoubiquit - ination, even after a 90-minute incubation period in which equivalent WT and missense variants had attained 100% FANCD2 monoubiquitination. The G750V variant showed near-normal levels of monoubiquitination of both FANCD2 and FANCI, indicating that this variant at protein level may not contribute toward pathogenicity.
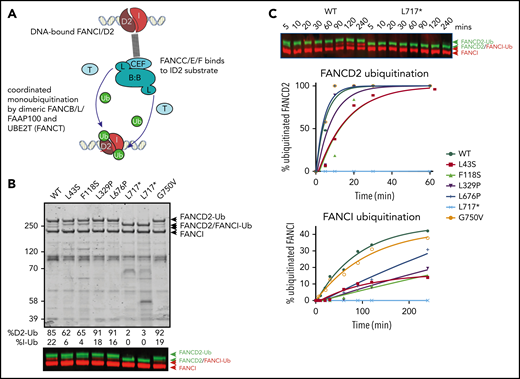
Effect of FANCB variants on ubiquitination activity of the FA core complex. (A) Schematic of in vitro ubiquitination reaction. (B) Results of an example monoubiquitination assay; Coomassie-stained sodium dodecyl sulfate–polyacrylamide gel electrophoresis gel to compare WT BL100 with BL100 with FANCB variants in 90-minute ubiquitination reaction. Western blotting of FANCD2 and FANCI reveals the extent of ubiquitinated protein, calculated from western blots using anti-StrepII-Fancd2 (green) or anti-Flag-FancI (red) (FANCI). Two different preparations of L717* complex are shown. (C) Example of time course experiment comparing monoubiquitination activity of WT and L717*-FANCB containing reactions by western blot (above) or quantified ubiquitinated forms of FANCD2 and FANCI. Similar results were obtained from n = 4 experiments.
Effect of FANCB variants on ubiquitination activity of the FA core complex. (A) Schematic of in vitro ubiquitination reaction. (B) Results of an example monoubiquitination assay; Coomassie-stained sodium dodecyl sulfate–polyacrylamide gel electrophoresis gel to compare WT BL100 with BL100 with FANCB variants in 90-minute ubiquitination reaction. Western blotting of FANCD2 and FANCI reveals the extent of ubiquitinated protein, calculated from western blots using anti-StrepII-Fancd2 (green) or anti-Flag-FancI (red) (FANCI). Two different preparations of L717* complex are shown. (C) Example of time course experiment comparing monoubiquitination activity of WT and L717*-FANCB containing reactions by western blot (above) or quantified ubiquitinated forms of FANCD2 and FANCI. Similar results were obtained from n = 4 experiments.
Individuals with FANCB pathogenic variants exhibit multiple congenital anomalies, early hematologic disease onset, and earlier death
Congenital malformations resembling VACTERL-H association were seen in 15 individuals with FANCB variants, with the predominant features being renal and limb abnormalities (observed in 13 individuals each; Table 1), which is consistent with previous observations.7,26 Interestingly, all 4 individuals (individuals 17, 18, 19, and 21) without VACTERL-H malformations carried missense variants. Twelve of 15 individuals with VACTERL-H association would be classified as VACTERL-PLUS in a nonsyndromic setting, according to a new classification system proposed by a EUROCAT study,27 because of other congenital anomalies outside the VACTERL spectrum. In general, developmental defects were multiple and severe for the individuals in our study; termination of pregnancy was elected for 3 IFAR individuals diagnosed prenatally and for 1 previously reported in the literature, and survival was limited to 0.033, 0.2, and 0.5 months for 3 other individuals.
The median survival time and mean age at onset of hematologic disease have been reported as 2428 and 7.6 years,2 respectively, among FA patients. Our data suggest that individuals with FA complementation group B have earlier death, with a median time to death of 104 months (n = 15, including individual 21; Table 1; supplemental Figure 8A), and earlier onset of hematologic disease, with a median and mean age of onset at 26 and 33.6 months, respectively (n = 11, including individual 21; Table 1; supplemental Figure 8B).
Seven individuals underwent hematopoietic stem cell transplantation (HSCT) for their hematologic disease; 2 died as a result of HSCT complications, whereas the other 5 are still alive at 108, 132, 132, 168, and 168 months of age. The survival for the remaining 4 with no HSCT was 1.1, 80, 104, and 136 months. In all, excluding the 7 individuals who underwent HSCT, the survival data, which were available for 8 individuals, revealed that no one survived beyond 136 months, and the median survival was 15.05 months.
To assess the genotype and phenotype correlation, we grouped variants based on the severity of structural alterations in the FANCB protein (WGD/truncation vs missense). The median time to death was earlier in the WGD/truncation variants group (15.05 months; range, 0.03-168 months) than in the missense group (104 months; range, 45-132 months), but was not statistically significant (log-Rank test, 0.5198; P = .4709; hazard ratio, 1.693; 95% confidence interval, 0.398-7.198; Figure 6A). Similarly, the median time to the hematologic disease was earlier in the WGD/truncation variant group (6.25 months; range, 0.25-48 months) than in the missense group (43 months; range, 12-102 months), but was also not statistically significant (log-rank test, 2.3962; P = .1216; hazard ratio, 3.058; 95% confidence interval, 0.669-13.973; Figure 6B). To assess the effect of residual function of missense variants on clinical outcome, we plotted outcomes of the activity of the identified variants and the clinical outcomes in the individuals with these variants (Figure 6C). The numbers of individuals are small; therefore, a definite conclusion cannot be drawn; however, clinical outcomes seem better with increased residual function of FANCB in individuals with missense variants when compared with those with WGD/truncation variants.
![Clinical severity correlates with in vitro residual FANCB function. (A) The median time to death was earlier in the WGD/truncation variant group (individuals 1, 2, 3, 4, 6, 11, 13, and 14) than in the missense group (individuals 16, 17, 18, 20, and 21) but was not statistically significant (log-rank test, 0.5198; P = .4709; hazard ratio [HR], 1.693; 95% confidence interval [CI], 0.398-7.198). Individuals excluded from this analysis are detailed in the supplemental methods. (B) The median time to hematologic disease was earlier in the WGD/truncation variant group (individuals 3, 6, 11, and 14) than in the missense group (individuals 16, 17, 18, 20, and 21) but was also not statistically significant (log-rank test, 2.3962; P = .1216; HR, 3.058; 95% CI, 0.669-13.973). Individuals excluded from this analysis are detailed in the supplemental methods. (C) Survival and onset of hematologic manifestations (heme onset defined as the onset of bone marrow failure, myelodysplastic syndrome, or leukemia, whichever came first) tends to correlate with residual function, such as mitomycin C sensitivity or quantity of FANCD2 foci. (D) A majority of the transcripts in the individual carrying c.1435T>G (predicted to express p.W479G) variant showed aberrant splicing and exon 7 skipping, which should result in an out-of-frame truncated protein. Although the expression level of c.1435T>G encoding the missense variant p.W479G was lower, its functional characteristics were near normal, correlating with the relatively milder phenotype in this individual. In contrast, the individual carrying splice site variant (c.1496+1G>A), which resulted in the only product that lacked exon 7, died on postnatal day 1. EV indicates vector-only control.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/18/10.1182_blood.2019003249/2/m_bloodbld2019003249f6.png?Expires=1769301542&Signature=PpjQl2ZQb53brWY7hylmhoaPQdZWPv6-Z2ISdhpTGbYLB6o-ousXqWmo~X7JdkJka7W0z3YG8CkD21z6r9qiam5biBaIHxtmTYFTIANLSTFcuKMvBwMrgGSUnjsoW6T9rQoOmseyE9ZpEBgnEkaDcK2qnmR0yylnvz3biMhDiQRhvHWm0oxSRU~y~6LrgPNMWXEeUnMoHdMDWDsVKtdBh5ooUpOdJ6x7yc533yfX19MSjYukdhYhyPMyrI~qbRImILtjBJB~YoHXeBdv1jICpUkMfNjGoFyrsxi4B4Vp9F-Xu-rLF0xsOoO7dIc5~C3ojdEqVJ-4dBSL1vygMaugag__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Clinical severity correlates with in vitro residual FANCB function. (A) The median time to death was earlier in the WGD/truncation variant group (individuals 1, 2, 3, 4, 6, 11, 13, and 14) than in the missense group (individuals 16, 17, 18, 20, and 21) but was not statistically significant (log-rank test, 0.5198; P = .4709; hazard ratio [HR], 1.693; 95% confidence interval [CI], 0.398-7.198). Individuals excluded from this analysis are detailed in the supplemental methods. (B) The median time to hematologic disease was earlier in the WGD/truncation variant group (individuals 3, 6, 11, and 14) than in the missense group (individuals 16, 17, 18, 20, and 21) but was also not statistically significant (log-rank test, 2.3962; P = .1216; HR, 3.058; 95% CI, 0.669-13.973). Individuals excluded from this analysis are detailed in the supplemental methods. (C) Survival and onset of hematologic manifestations (heme onset defined as the onset of bone marrow failure, myelodysplastic syndrome, or leukemia, whichever came first) tends to correlate with residual function, such as mitomycin C sensitivity or quantity of FANCD2 foci. (D) A majority of the transcripts in the individual carrying c.1435T>G (predicted to express p.W479G) variant showed aberrant splicing and exon 7 skipping, which should result in an out-of-frame truncated protein. Although the expression level of c.1435T>G encoding the missense variant p.W479G was lower, its functional characteristics were near normal, correlating with the relatively milder phenotype in this individual. In contrast, the individual carrying splice site variant (c.1496+1G>A), which resulted in the only product that lacked exon 7, died on postnatal day 1. EV indicates vector-only control.
Clinical severity correlates with in vitro residual FANCB function. (A) The median time to death was earlier in the WGD/truncation variant group (individuals 1, 2, 3, 4, 6, 11, 13, and 14) than in the missense group (individuals 16, 17, 18, 20, and 21) but was not statistically significant (log-rank test, 0.5198; P = .4709; hazard ratio [HR], 1.693; 95% confidence interval [CI], 0.398-7.198). Individuals excluded from this analysis are detailed in the supplemental methods. (B) The median time to hematologic disease was earlier in the WGD/truncation variant group (individuals 3, 6, 11, and 14) than in the missense group (individuals 16, 17, 18, 20, and 21) but was also not statistically significant (log-rank test, 2.3962; P = .1216; HR, 3.058; 95% CI, 0.669-13.973). Individuals excluded from this analysis are detailed in the supplemental methods. (C) Survival and onset of hematologic manifestations (heme onset defined as the onset of bone marrow failure, myelodysplastic syndrome, or leukemia, whichever came first) tends to correlate with residual function, such as mitomycin C sensitivity or quantity of FANCD2 foci. (D) A majority of the transcripts in the individual carrying c.1435T>G (predicted to express p.W479G) variant showed aberrant splicing and exon 7 skipping, which should result in an out-of-frame truncated protein. Although the expression level of c.1435T>G encoding the missense variant p.W479G was lower, its functional characteristics were near normal, correlating with the relatively milder phenotype in this individual. In contrast, the individual carrying splice site variant (c.1496+1G>A), which resulted in the only product that lacked exon 7, died on postnatal day 1. EV indicates vector-only control.
Discussion
FANCB represents 2.2% of all and 4.0% of male FA individuals who were genotyped as part of the IFAR, and we describe here 16 FANCB families, each with distinct variants (Figure 1; supplemental Figure 1; Table 1). Other previously reported FANCB variants were also found only in single families,5,8,20,21,29-34 suggesting an absence of founder mutations. This is likely due to the short lifespan of male carriers and the severity of symptoms, which probably extends to infertility, as in Fancby/− mice.35 The nature of the variants included large deletion/duplication, missense, nonsense, splice junction, and indel variants and an unusual combination of overlapping de novo missense and indel variants. Deletions/duplications accounted for 20% to 30% of all variants in this and other studies.5,8,31 Deletions range from small (∼3 kb, removing a single exon5 ) to large (520-590 kb, eliminating the entire FANCB gene along with flanking genes; individuals 1-3). Whether the concurrent deletion of MOSPD2 and GLRA2 observed in individuals 1 to 3 contributed to the severe phenotype in these individuals cannot be determined because of small sample sizes. Identification of these FANCB variants relies on methods for identifying large deletions, such as aCGH.36 Confirmation of the de novo status of FANCB variants, by eliminating the possibility of mosaicism in the parents, requires specialized techniques, such as deep sequencing, which is a robust method to detect unstable variants at low frequencies. In our cases, we found no evidence for mosaicism in probands with de novo or inherited variants or heterozygous carrier mothers/siblings (supplemental Tables 2 and 5). However, for individual 15, who carried 2 overlapping de novo variants, we observed that the 2 variants existed at varying frequencies in DNA of peripheral blood and the LCL. The reduced fraction of the c.2249G>T (G750V) variant in blood, and to an even greater extent in the LCL, suggests that cells carrying indel variant transcript have a selective growth advantage. This could be due to the unstable nature of the missense variant that was observed both in LCLs derived from individual 15 and in FANCB-null cells overexpressing G750V (Figure 3A; supplemental Figure 3). These findings suggest that the missense variant originated before the indel variant. Observations that transcripts differ from predictions based on genomic variants demonstrate the importance of including transcript analysis as part of comprehensive molecular diagnosis.
Exon skipping played a role in our FANCB FA cohort, and the level of retained missense transcript (and hence full-length FANCB protein) also correlated with clinical features. The exon 7 nonsynonymous variant, c.1435T>G (p.W479G), in individual 19 induced exon 7 skipping in ∼85% of transcripts, reducing the contribution of the predicted nonsynonymous protein variant p.W479G to <15% (Figure 2B-E). Functional studies revealed that the W479G FANCB protein retains substantial levels of FANCD2 ubiquitylation, foci formation, and ability to rescue null cell viability (Figure 3). In contrast, individual 4, with the splice donor variant c.1496+1G>A, generated the same aberrant transcript with exon 7 skipped, but in 100% of transcripts (Figure 2B-E). Individual 4 died 1 day after birth, whereas individual 19 lived until 120 months, dying as a result of HSCT complications (Figure 6D). We and others have reported earlier that aberrant splicing is associated with certain synonymous, nonsynonymous, and nonsense variants in FA genes,17,31,37 indicating that quantitative RNA analysis should be included to evaluate the molecular consequence of a genomic variant, particularly in the absence of clearly pathogenic alterations, but an apparent FA phenotype.
Little is known about the actual function of FANCB protein within the FA core complex. Previous studies have shown that the protein is stable for purification only as a recombinant heterodimer with FAAP100.12 Imaging of the FANCB/FAAP100 complex by electron microscopy suggests that the proteins form an intricately intertwined complex containing 2 of each subunit. We hypothesized that this unusual arrangement could place 2 FANCL molecules, each in close proximity to the ubiquitination sites of the subunit of the FANCD2/FANCI heterodimeric substrate.14 Interestingly, we find that many variants in FANCB have a greater effect on FANCI monoubiquitination and only mild or no effect on FANCD2 monoubiquitination in vitro. It is possible that FANCB, as the central dimerization mediator, coordinates the sequential monoubiquitination of FANCD2 and FANCI. Importantly, one of the longest reported truncations in FANCB completely lacks any detectable monoubiquitination activity, even though it retains association with FANCL, FAAP100, and the remainder of the FA core complex. This suggests that the C-terminus of FANCB has an essential role in the catalytic activity of the entire FA core complex. From this finding, we predict that even small FANCB truncations would be pathogenic.
We also identified 2 variants in the N-terminus of FANCB that cause an abrogation of interaction with the remainder of the FA core complex. The ∼60% to 80% reduction in core complex association is sufficient to reduce the kinetics of FANCD2 and FANCI monoubiquitination by a similar rate. In a cellular context, where FA core complex concentrations are much more limiting (∼0.1 ppm in Protein Abundance Database),38 such a reduced rate of association is likely to have an even greater effect on FANCD2 monoubiquitination. Together these findings suggest that a predicted β-propeller in the N-terminus of FANCB mediates association with the FA core complex, whereas the remainder of the protein mediates the dimerization and catalytic activity of FANCL E3 RING ligase.
In this study, we report that individuals with FANCB variants seem to have earlier onset of bone marrow failure and experience severe congenital anomalies more frequently than the general FA individuals. Individuals with congenital anomalies resembling VACTERL-H association were frequently in the VACTERL-PLUS subtype because of other congenital anomalies outside the VACTERL spectrum; no VACTERL-STRICT subtype was observed in our cohort.27 It is noteworthy that presence of FA or VACTERL-H was an exclusion criterion in the EUROCAT study. FA should be ruled out in cases of VACTERL-PLUS.
We also observe phenotypic variation among individuals with different FANCB pathogenic variants. Individuals with WGD/truncation variants in FANCB had more severe phenotypes than individuals with missense variants, although our analyses were limited by small sample sizes (Table 1; Figure 6A-B). Although it is ideal to perform the time to death analysis with HSCT status as a time-varying covariate, we did not have a large enough sample size to draw a meaningful conclusion from such analysis. Therefore, it was not reported. Results from all the functional assays of missense variants, both from cellular (Figure 3) and in vitro assays (Figures 4 and 5) as well as transcript analysis, are summarized in Table 2. All of the missense variants tested were found to be pathogenic by ≥1 assays. In particular, compared with WT FANCB, a majority of variants showed relatively lower levels in the nucleus (supplemental Figure 6), which is also a characteristic of pathogenic FANCA missense variants.37 Together, the functional assays revealed a spectrum of activity, and the cell-based and protein-based assays broadly agreed on the extent of loss of function. The residual effect of each missense variant on cell viability and FANCD2 foci formation seemed to have some correlation with hematologic disease onset and overall survival in the individuals with FANCB missense variants (Figure 6C).
Because FANCB is X-linked and, to date, restricted to male cases, each affected individual carries only 1 deleterious FANCB allele and generally displays a more severe form of FA with earlier hematologic onset, shorter survival, and more frequent VACTERL-H association. Our study highlights how complementary approaches to variant analysis converge to provide meaningful new knowledge of both FANCB protein function and the etiology of FANCB-associated FA.
For original data, please e-mail the corresponding authors.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the individuals and families who participated in this study. They also thank K. J. Patel for the FANCE antibody.
This work was supported by grants from the National Institutes of Health (NIH) National Heart Lung and Blood Institute (R01 HL120922) (A.S.), National Cancer Institute (R01 CA204127) (A.S.), National Center for Advancing Translational Sciences (UL1 TR001866) (M.J., C.S.J., R.V., and A.S.), American Society of Hematology Scholar Award (M.J.), the National Health and Medical Research Council, Australia (APP1123100 and APP1145391) (A.J.D.), Cancer Australia (GNT1125750) (A.J.D.), and the Victoria government IOS program (A.J.D.). A.S. is a Howard Hughes Medical Institute Faculty Scholar. A.J.D. is a Victorian Cancer Agency midcareer fellow. R.R.-B., F.X.D., D.C.K., and S.C.C. acknowledge support from the Intramural Research Program of the NIH National Human Genome Research Institute.
Authorship
Contribution: M.J., R.R.-B., S.v.T., F.P.L., V.M., and W.T. performed experiments; M.J., R.R.-B., S.v.T., F.X.D., S.C.C., and A.J.D. analyzed results; R.R.-B., D.C.K., F.X.D., and S.C.C. collected and analyzed the genomic sequence; M.J., R.R.-B., and A.J.D. made the figures; R.O.R., F.P.L., and A.S. managed the IFAR; A.D.A. established the IFAR, obtained follow-up clinical information, and edited the paper; C.S.J. and R.V. performed statistical analysis of clinical data; M.J., R.R.-B., A.J.D., A.S., and S.C.C. designed the research and wrote the paper; and R.O.R., P.A.M., F.P., and C.D. collected clinical presentations of some of the individuals.
Conflict-of-interest disclosure: A.S. receives research funding from Rocket Pharmaceuticals for an unrelated project. The remaining authors declare no competing financial interests.
Correspondence: Andrew J. Deans, Genome Stability Unit, St Vincent’s Institute of Medical Research, 9 Princes St, Fitzroy, VIC 3065, Australia; e-mail: adeans@svi.edu.au; Agata Smogorzewska, 1230 York Ave, Box 182, New York, NY 10065; e-mail: asmogorzewska@rockefeller.edu; and Settara C. Chandrasekharappa, NIH/NHGRI/CGCGB, 50 South Dr, Bldg 50, Room 5232, Bethesda, MD 20892-8004; e-mail: chandra@mail.nih.gov.

![Clinical severity correlates with in vitro residual FANCB function. (A) The median time to death was earlier in the WGD/truncation variant group (individuals 1, 2, 3, 4, 6, 11, 13, and 14) than in the missense group (individuals 16, 17, 18, 20, and 21) but was not statistically significant (log-rank test, 0.5198; P = .4709; hazard ratio [HR], 1.693; 95% confidence interval [CI], 0.398-7.198). Individuals excluded from this analysis are detailed in the supplemental methods. (B) The median time to hematologic disease was earlier in the WGD/truncation variant group (individuals 3, 6, 11, and 14) than in the missense group (individuals 16, 17, 18, 20, and 21) but was also not statistically significant (log-rank test, 2.3962; P = .1216; HR, 3.058; 95% CI, 0.669-13.973). Individuals excluded from this analysis are detailed in the supplemental methods. (C) Survival and onset of hematologic manifestations (heme onset defined as the onset of bone marrow failure, myelodysplastic syndrome, or leukemia, whichever came first) tends to correlate with residual function, such as mitomycin C sensitivity or quantity of FANCD2 foci. (D) A majority of the transcripts in the individual carrying c.1435T>G (predicted to express p.W479G) variant showed aberrant splicing and exon 7 skipping, which should result in an out-of-frame truncated protein. Although the expression level of c.1435T>G encoding the missense variant p.W479G was lower, its functional characteristics were near normal, correlating with the relatively milder phenotype in this individual. In contrast, the individual carrying splice site variant (c.1496+1G>A), which resulted in the only product that lacked exon 7, died on postnatal day 1. EV indicates vector-only control.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/18/10.1182_blood.2019003249/2/m_bloodbld2019003249f6.png?Expires=1769301543&Signature=drFLdI0cLxmhbo0CJe6V7cB6q19NCN3LNbNYKnQECzyquMgK18NJrYT5Gvcm9uKJPq-4QEVI-udMewCYKhfBoeBWZW9TzzmEGQqt6pva7MHjVVPrNCw7nYyLuEH~HPqSlBDkEHX9vkObaPHrdtyevjRhoCyo3HS7UaTOKfpYNbvTQp1-gli10BTkg9OhY~oVlwgaQrmpeAN5VfUEDYNTtNXYfBX3TqZ3sL3KY59RwnVW-CuxkZyVDnHPxHkECf26CLqeQuyg3KhZGtYv7fgs8CpfYOibs6ry6mh4EHZ~HahsTvp6~rz5JI3Alygl2-JTnTbVjLtoLrl-f2jaZHt0Gw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
