Abstract
Common variable immune deficiency (CVID) is one of the most common congenital immune defects encountered in clinical practice. The condition occurs equally in males and females, and most commonly in the 20- to 40-year-old age group. The diagnosis is made by documenting reduced serum concentrations of immunoglobulin G (IgG), IgA, and usually IgM, together with loss of protective antibodies. The genetics of this syndrome are complex and are still being unraveled, but the hallmarks for most patients, as with other immune defects, include acute and chronic infections of the sinopulmonary tract. However, other noninfectious autoimmune or inflammatory conditions may also occur in CVID, and indeed these may be the first and only sign that a significant immune defect is present. These manifestations include episodes of immune thrombocytopenia, autoimmune hemolytic anemia, or neutropenia, in addition to splenomegaly, generalized or worrisome lymphadenopathy, and malignancy, especially lymphoma. These issues commonly bring the patient to the attention of hematologists for both evaluation and treatment. This article discusses 3 cases in which patients with CVID had some of these presenting issues and what hematology input was required.
“Common variable immune deficiency” (CVID) is the umbrella name for a collection of hypogammaglobulinemia syndromes in which low levels of serum immunoglobulin G (IgG), IgA, and/or IgM, are accompanied by defects of antibody production. The name was first applied to this immune defect in 1971 in order to attempt to separate these patients from those with clinically better-defined syndromes such as X-linked agammaglobulinemia, Wiskott-Aldrich syndrome, hyper-IgM syndromes, or other defects with more unified clinical descriptions and often obvious Mendelian inheritance.1 Although considered to be a genetic defect, a puzzling feature has always been that the diagnosis is most commonly made in adults between ages 20 and 40 years; however, in all series, 20% are younger, and a number of much older adults are first diagnosed in later years.2,3 Although the percentage of patients with monogenetic defects associated with CVID has increased to about 20% to 25%, understanding of the pathogenesis of this syndrome is based on many immunological studies performed in cohorts of various sizes from medical centers with different interests. Many studies address the lack of long-lived specific antibody responses, whereas other studies have investigated the reasons for and deficits produced by abnormal cellular immune pathways. The incidence of CVID is estimated to be between 1:25 000 and 1:50 000 in white individuals, but it is apparently less common in Asian and African American individuals. It is equally prevalent in males and females. In all studies of CVID, a delay in making the diagnosis (range from 0 to 61 years) has been noted, with 20% of patients being diagnosed with CVID more than 15 years after the onset of cardinal symptoms. In the European Society for Immune Deficiency (ESID) data set of 388 patients, the mean diagnostic delay was 7.46 years, and the median was 5 years.
Although exact definitions vary, consensus groups have defined CVID as requiring a marked decrease of IgG (≥2 standard deviations below mean for age) together with a marked reduction in one or both of the isotypes IgA (usually) or IgM, as well as poor or absent antibody responses to vaccines or microbial illnesses.1,4 Antibody responses are commonly examined by determining IgG responses to 2 or more protein vaccines (tetanus or diphtheria toxoids; Haemophilus conjugate; measles, mumps, and rubella vaccines) and also by lack of responses to pneumococcal polysaccharide vaccines to prove and define the extent of the deficiency. Although extensive antibody testing is not as important for subjects with very low serum IgG (potentially 150-200 mg/dL or less), those with higher levels of serum IgG (450-600 mg/dL), and especially those with only minimally reduced serum IgA, require more extensive evaluation of specific antibody production. Young children, generally aged <4 years, are not usually given the diagnosis of CVID, because other causes are more likely in this age group. However, if the immune defect persists and no other causes are found, the CVID term can be used. Subjects with IgG deficiency alone are best segregated from CVID into a separate category because the immune and clinical phenotypes differ significantly.5 Patients with quite reduced numbers of CD4+ T cells, especially naïve T cells, need special consideration because these individuals are more likely to have a separable, genetically defined combined immune defect.6
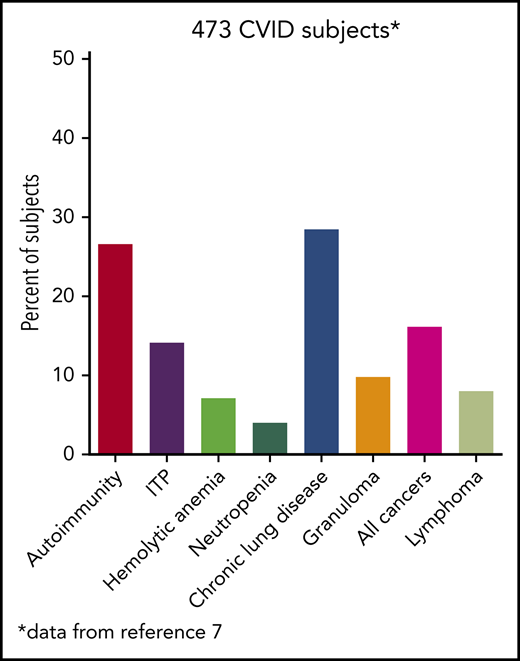
Other laboratory features found useful in evaluating the patient with CVID are the numbers and phenotypes of peripheral blood B cells. CD27+ memory B cells, but especially IgD-CD27+ isotype-switched memory B cells, are decreased.7 Very low levels of isotype-switched memory B cells are significantly associated with autoimmunity, granulomatous disease, hypersplenism, lymphoid hyperplasia, and possibly chronic lung disease. Other B-cell markers include increased numbers of CD21lo B cells, also associated with autoimmunity,7 and increased numbers of transitional B cells, suggesting continued immaturity.
The hallmarks of CVID, as for other immune defects, include acute and chronic infections of the sinopulmonary tract in most patients. Other clinical manifestations are quite broad and include autoimmunity; splenomegaly with lymphadenopathy; and, in some, an increased incidence of various cancers, especially lymphoma. These issues commonly bring the patient to the attention of hematologists. A rather common presentation of CVID is a patient with recurrent autoimmune cytopenias, either immune thrombocytopenia (ITP), autoimmune hemolytic anemia (AIHA), or occasionally both. In aggregate, different forms of autoimmunity occur in up to 30% of subjects with CVID.2,8
Clinical case 1: recurrent AIHA
Case description
A 46-year-old male contractor who smoked but was previously healthy had acute AIHA. He was treated with prednisone and recovered. This occurred again the following year, and he required blood transfusions. Four years later, AIHA again occurred; he developed a clot in his aorta with thrombosis of his splenic artery, resulting in a splenic infarct. He had a splenectomy and recovered uneventfully. The following year, he developed pneumonia with a large posterior empyema; Streptococcus pneumoniae was cultured (Figure 1). The collection was drained, complicated by a collapsed lung, but he recovered. Six months later, he had a second bacterial pneumonia, and immunoglobulin was measured. These tests showed remarkably low levels of serum immunoglobulins: IgG, 71 mg/dL (normal range, 639-1349); IgA, 6 mg/dL (80-350); and IgM, 15 mg/dL (45-250). He had no protective titers of antibody to any vaccines. Intravenous immunoglobulin replacement was instituted, and the patient experienced no further episodes of either infection or autoimmunity.
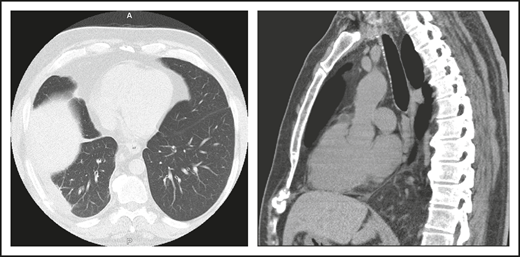
(A) The patient in clinical case 1 with several episodes of severe autoimmune hemolytic anemia was only diagnosed with CVID after he had bacterial pneumonia and developed an empyema. (B) Lateral view shows the posterior empyema collection.
(A) The patient in clinical case 1 with several episodes of severe autoimmune hemolytic anemia was only diagnosed with CVID after he had bacterial pneumonia and developed an empyema. (B) Lateral view shows the posterior empyema collection.
Hematologic autoimmunity
This patient had profound hypogammaglobulinemia and yet was still capable of producing anti–red blood cell antibodies, one of the mysteries of CVID, because sufficient levels of autoantibodies leading to clinical illness can still appear. Autoimmunity, which may be recurrent as in this case, occurs in at least 30% of patients with CVID and may be the first manifestation of the immune defect in patients, some of whom have never had a significant infection.3,9,10 The most common is immune-mediated thrombocytopenia; less commonly, AIHA or, even more uncommonly, autoimmune neutropenia is seen (Table 1). Among 311 patients with CVID in the French registry, 55 (18%) patients had an autoimmune cytopenia.11 Of 990 patients with CVID in the National Institutes of Health–supported United States Immunodeficiency Network (USIDNET) Registry, which is managed by the Immune Deficiency Foundation, 10.2% had an autoimmune cytopenia. ITP was diagnosed in 7.4%, AIHA in 4.5%, and autoimmune neutropenia in 1%8 ; several had Evans syndrome. Patients with autoimmunity are also likely to have one or more other noninfectious complications: lymphoproliferation, granulomatous disease, liver dysfunction, interstitial lung diseases, enteropathy, or lymphoma.8
Autoimmunity is not restricted to hematologic cells; rheumatologic complications occur in perhaps 10% of patients. Other conditions include pernicious anemia, primary biliary cirrhosis, thyroiditis, sicca syndrome and Sjögren syndrome, systemic lupus, vitiligo, and alopecia.11 In subjects with autoimmune cytopenias, a central observation is that there is a significantly increased proportion of CD21lo B cells.9,11 Romberg et al, investigating the pathology of lymph nodes of these subjects, reported irregularly shaped hyperplastic germinal centers, whereas germinal centers were scarce and smaller in patients without this history. This hyperplasia was accompanied by an increase in numbers of circulating follicular helper T cells, with both decreased regulatory T-cell numbers and decreased function.12
Why is autoimmunity so common in CVID? The answers to this question are complex and likely include many causes: failure to counterselect polyspecific/self-reactive clones that arise in the bone marrow in even healthy humans; genetic mutations in CVID that foster autoimmunity; immature B-cell development7,13 ; and high levels of factors in serum that lead to B-cell proliferation, including BAFF (B-cell activating factor) and APRIL (a proliferation inducing ligand), both of which drive the immature B cells.14,15 Additional reasons for autoimmunity in CVID include loss of regulatory T cells16 and chronic antigen stimulation.
Clinical case 2: splenomegaly/lymphadenopathy and a wrong diagnosis
Case description
A second common reason for a patient to come to the attention of hematologists is the finding of an enlarged spleen, especially in an adult with lymphadenopathy. This was the reason in patient 2, who is now a 46-year-old male professional dog walker. At the age of 31, he was found to have an enlarged spleen together with cervical and axillary lymphadenopathy. Hematology was consulted, and the lymph node was biopsied; pathology revealed that he had noncaseating granuloma. He was then referred to pulmonology and, with this evidence, was diagnosed with sarcoidosis and treated with steroids for some months, but he stopped the treatment. Although he had no obvious respiratory symptoms over the following 15 years, he had a slow decline in lung function, and chest computed tomography showed hilar lymphadenopathy, ground-glass opacities with nodules, and bronchiectatic changes (Figure 2). In 2018, he had acute bacterial pneumonia, became concerned, and found on the internet that granulomata in the lungs and an enlarged spleen might be related to low serum immunoglobulin levels. He asked that this be tested; when this test was performed, he had striking low serum immunoglobulins: IgG, 97 mg/dL; IgA, <5 mg/dL; IgM, 27 mg/dL. However, within the month, he was hospitalized for pneumonia due to metapneumovirus. The diagnosis of CVID was finally made, and immunoglobulin treatment was started. The patient’s spleen remains large.
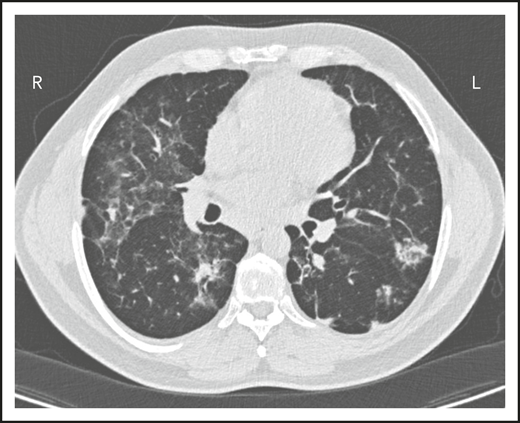
The patient in clinical case 2 had a slow decline in lung function due to his immune defect, but this was assumed to be due to sarcoidosis. Chest computed tomography showed ground-glass opacities with numerous nodules, hilar lymphadenopathy, and bronchiectatic changes. With granuloma on biopsy, these changes were consistent with granulomatous lymphocytic interstitial lung disease.
The patient in clinical case 2 had a slow decline in lung function due to his immune defect, but this was assumed to be due to sarcoidosis. Chest computed tomography showed ground-glass opacities with numerous nodules, hilar lymphadenopathy, and bronchiectatic changes. With granuloma on biopsy, these changes were consistent with granulomatous lymphocytic interstitial lung disease.
Splenomegaly and lymphadenopathy
An enlarged spleen, as noted in this case, sometimes with cervical and hilar lymphadenopathy, is quite common in CVID and may be the only physical finding. In the ESID registry, splenomegaly was reported in 26% of subjects with CVID.2 It was noted frequently at the time of diagnosis of CVID and, in these cases, was commonly associated with either a history of autoimmunity or the diagnosis of granulomatous disease, usually in the lungs or lymph nodes, or both (P = .01). Another common association of splenomegaly in CVID is an enlarged liver.10
In CVID, biopsies of lymph nodes usually show atypical or reactive hyperplasia, with or without preservation of germinal center boundaries; granulomatous infiltrations are found in some. There is typically a lack of plasma cells in lymphoid tissues (eg, lymph nodes, the gastrointestinal tract, and bone marrow) in CVID17 ; this sometimes provides a first clue that an immune defect is present. In most cases, for splenomegaly or enlarged nodes, no specific treatment is required. Spleens can be massive and yet not cause clinical symptoms. Splenectomy is almost always avoidable, unless there is clinically marked hypersplenism, uncontrollable autoimmunity (which can usually be treated by rituximab as discussed in the Treatment section), or a real possibility of lymphoma (see the Treatment section).
Granuloma in tissues
Localized or systemic granulomatous disease, sometimes erroneously called “sarcoidosis,” occurs in between 8% and 22% of subjects with CVID.18,19 For unclear reasons, many of these patients have also had ITP or AIHA. In the patient in clinical case 2, although he did not have characteristic lung infections such as pneumonia for many years, the diagnosis of “sarcoid” retarded the recognition of the underlying immune defect; this confusion has repeatedly been observed, and the delay in diagnosis can be for more than a decade, as it was in this case.19 Organisms must be sought but are very rarely found. In CVID, the granulomatous changes are usually found in lungs, lymph nodes, or spleen, but the skin, liver, bone marrow, kidney, gastrointestinal tract, and brain can also be involved. In the lungs, an intense lymphoid infiltration accompanies the granulomas, leading to what is termed “granulomatous lymphocytic interstitial lung disease.”18 For unclear reasons, patients with granulomatous disease are likelier to have ITP or AIHA than those with CVID, who do not have this pathology and thus require the attention of hematologists.
Clinical case 3: concern for lymphoma
Case description
A third common reason for hematologists to see patients with CVID is a suspicion of lymphoma. Patient 3 is now a 47-year-old computer executive who came for a second opinion to the immunology department. He had had numerous sinopulmonary infections in childhood and then 2 episodes of shingles in his 30s. At the age of 36, he had acute AIHA treated with steroids. He then had an episode of cellulitis, and, with this, serum immunoglobulins were tested, which showed quite low levels: IgG, 186 mg/dL; IgA, 9 mg/dL; and IgM, 24 mg/dL. At the same time, his spleen was quite large. He was diagnosed with CVID, and immunoglobulin therapy was initiated. Owing to the enlarged spleen, now at 19 cm, a bone marrow biopsy was performed, and he was ultimately seen at another center at the age of 42, when the diagnosis of marginal zone lymphoma was made on the basis of bone marrow findings. He was treated with 6 cycles of rituximab and bendamustine. However, his splenomegaly persisted, and 3 years later, he sought an opinion at another medical center, where he was told that he did not have lymphoma on the basis of National Cancer Institute review of his bone marrow. Now, 5 years later, he has been well while receiving intravenous immunoglobulin, but he continues to have an enlarged spleen. There are no signs of lymphoma (Figure 3).
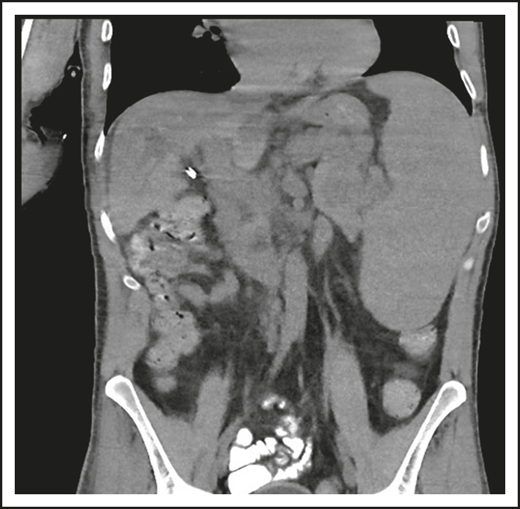
The patient in clinical case 3 was concerned because after he received chemotherapy for a presumed lymphoma, his spleen remained large. However, this is typical in patients with CVID.
The patient in clinical case 3 was concerned because after he received chemotherapy for a presumed lymphoma, his spleen remained large. However, this is typical in patients with CVID.
Lymphoma and cancer
There is clearly an overall increase in the incidence of malignancy in CVID, especially in non- Hodgkin lymphoma, as shown in a number of reports (Table 2). In an Italian study, 20.7% of subjects with CVID followed over 4 decades developed various forms of cancer, including lymphoma.20 For 176 subjects with CVID in another study, the observed-to-expected ratio for lymphoma was 12.1, a clear increase in incidence. Cancers of the stomach have also been noted in most series and may be the cause of death.21,22 In the United States, for a cohort of 476 patients at one center, 39 patients (8.2%) had a lymphoid malignancy; all were B cells in type, and non-Hodgkin B-cell lymphomas were the most common. Some of these were further classified into mucosa-associated lymphoid tissue (MALT) lymphoma, marginal zone lymphoma, and T-cell–rich B-cell Epstein-Barr virus (EBV)-associated lymphoma. There were also 4 cases of Hodgkin disease.3 One patient in this group was initially diagnosed with MALT lymphoma; however, his pathology was also reviewed at the National Cancer Institute, and in his case, the diagnosis was reclassified as monoclonal B lymphocytosis. In the USIDNET Registry, in 1259 patients with CVID, there was also a higher incidence of lymphoma (n = 16 in men and n = 21 in women) than the expected lymphoma incidence of 1.9 in men (P < .001) and 3 in women (P < .001).23
Although lymphomas do occur in CVID, it must be emphasized that careful distinction between malignant and nonmalignant lymphoid proliferation in CVID is not simple, and as pointed out previously, clonal expansions occur in nonmalignant nodes, tissues, and blood in patients with CVID.24,25 When lymphomas do appear in CVID, they can be extranodal, usually B cell in type, EBV negative, and more common in subjects in the 4th to 7th decades of life. Lymphomas in CVID are not usually diagnosed by bone marrow biopsy; positron emission tomographic scans in CVID often demonstrate splenic enlargement and known lymphoid hyperactivity but may help point to a site for biopsy. A number of cases of marginal zone (MALT) lymphomas have been reported,26 in some cases related to Helicobacter pylori. Lymphoma may be more likely to arise in subjects with preexisting polyclonal lymphoproliferation, as shown for 10 cases in 334 subjects with CVID extracted from the ESID Registry. In this study, a higher baseline serum IgM in CVID was correlated with both lymphoid hyperplasia and lymphoma.10 These data were also confirmed in a US cohort of 476 subjects with CVID.3 However, as shown in clinical case 3, although lymphoid malignancies are increased in CVID, the diagnosis requires careful review. Potentially due to the intrinsic B-cell defect(s), a constricted B-cell repertoire with clonality is common in CVID,24 and as previously pointed out, distorted germinal centers can be found in subjects with CVID who do not have lymphoma. Without careful immunophenotypic and molecular studies, these can be mistaken for lymphoma.
CVID and genetics
Because of the late onset of clinical symptoms and striking heterogeneity, CVID has often been assumed to be a polygenic syndrome. However, whole-exome and whole-genome analyses have now revealed an increasing number of monogenic causes of the CVID phenotype. The first were autosomal recessive genes, but an increasing number of autosomal dominant genes with variable penetrance have been documented, accounting for up to 30% of patients in some reports.27 The genes identified reflect the complex requirements of B-cell antigen signaling, activation, survival, migration, and maturation to the plasma cell stage. The main genes linked to this syndrome are discussed briefly here and are outlined in Table 3. Autosomal recessive genes include the gene encoding the T-cell surface receptor, inducible T-cell costimulator (ICOS)28 ; the BAFF receptor29 ; and the genes for the B-cell receptor complex associated proteins CD19, CD20, CD21, CD81, and CD27.30 Mutations in TACI (calcium-modulating cyclophilin ligand interactor) are found in 8% to 10% of patients with CVID, usually in the heterozygous state, suggesting either dominant-negative effect or haploinsufficiency.31,32 However, it is clear that TACI variants can also be found in first-degree relatives and some healthy individuals, making these more of an association with CVID than a definitive cause. Clinically, patients with these mutations have a propensity to autoimmune manifestations and lymphoid hyperplasia, potentially due to lack of normal mechanisms of establishing tolerance.33 Although Ikaros proteins are lymphoid-restricted zinc finger transcription factors considered to be master regulators of lymphocyte differentiation in general, autosomal dominant mutations in the IKZF1 gene lead to CVID.34 Another emerging theme is that mutations in genes involved in immune regulation may also lead to a clinical picture of CVID. These include autosomal recessive mutations in LRBA (lipopolysaccharide-responsive beige-like anchor) protein35 in which patients have recurrent infections, autoimmunity, and almost autoimmune/lymphoproliferative-like features in some and severe inflammatory bowel disease in others.27 Similar to defects of LRBA, but with more variable penetrance, are the heterozygous autosomal dominant mutations in CTLA4, another member of the ICOS/B7 (CD80) family.36 (These defects are mechanistically closely related because LRBA serves as a chaperone that controls CTLA4 expression.) Patients may have autoimmunity, lymphoid hyperplasia, enteropathy, and granulomatous infiltrative lung disease.37 Another set of mutations that may produce CVID (or more of a hyper-IgM phenotype) comprises autosomal dominant mutations in phosphatidylinositol 3-kinase, in particular the catalytic subunit (PIK3CD). In addition to these, mutations in transcription factors of the NF-κB family are now being described in increasing numbers of subjects with CVID. Autosomal dominant defects in the NFKB1 subunit may be the most common, leading to antibody deficiency, respiratory infections, unusual infections, autoimmunity, and lymphoproliferative disease.28,38 In addition to the NFKB1 gene, heterozygous autosomal dominant mutations in NFΚB2 may also lead to early-onset hypogammaglobulinemia with recurrent infections and autoimmunity in some, but in others also endocrine abnormities.39
A more important question to answer is which patients with CVID might benefit from genetic studies, and, if so, whether targeted panels that dissect B-cell defects or whole-exome sequencing methods should be used. Although no definitive answers are possible, subjects with difficult-to-control autoimmunity, lymphocytic infiltrations in lungs or organs, and granulomatous disease or lymphomas may benefit most from these studies because pharmacologic or biologic treatments may be usefully applied.
Treatment
Replacement immunoglobulin
The standard of care in CVID treatment is replacement immunoglobulin via either the IV or subcutaneous route.40 Immunoglobulin greatly reduces the number of bacterial infections41 and increases life expectancy, and it reduces the frequency of autoimmune cytopenias. However, it does not appear to protect against the other noninfectious inflammatory complications, such as lung disease, granulomatous disease, enteropathy, or the development of cancer or lymphoma, which require additional attention and the potential use of biologics. Antibiotics are needed for acute treatment and, in many cases, also on a chronic basis as prophylaxis.3,10
Treatment of autoimmune cytopenias
Acute treatment of AIHA (or ITP) in CVID usually produces a response to oral (dexamethasone or prednisone) or IV steroids (1 g methylprednisolone), with or without large doses of immunoglobulin, followed by moderate doses of oral steroids tapered over several weeks or more. In addition, or alternatively, rituximab in standard doses has proved effective for more refractory or recurrent ITP and/or AIHA, but this is reserved (and safest) for subjects receiving immunoglobulin therapy.42 For subjects with recurrent cytopenias, chronic treatment with IV or subcutaneous immunoglobulin reduces the likelihood of additional episodes43 ; however, a recent publication noted that a consistent IgG level ≥700 mg/dL was required for this benefit to be observed.43 Splenectomy is to be avoided in CVID because severe infections have occurred, as we and others have shown, although this is not found in all reports.44 However, splenomegaly can cause physical discomfort and/or significant hypersplenism; thus, splenectomy is sometimes required. Fortunately, this does not appear to increase mortality in CVID if adequate immunoglobulin replacement is used.3,44 Although autoimmune cytopenias such as ITP and AIHA may respond to high-dose immunoglobulin and/or steroids, added therapy such as rituximab has been widely used and often provides long-term benefit.42 In a retrospective multicenter study, rituximab was highly effective in 33 patients with CVID-associated immune cytopenias, leading to an initial response rate of 80% and a sustained response rate of 50% after a mean follow-up of 39 ± 30 months after the first administration. Note that because immunoglobulin infusions can interfere with the effectiveness of rituximab, an interval is needed between these treatments. Other more recent options for thrombocytopenias include thrombopoietin receptor agonists, which have also shown benefit.45
Treatment of granulomatous disease
The best treatment of the granulomatous form of CVID is not established and depends largely on the sites of involvement. When the lungs are involved, and especially when lung function is impaired, morbidity and mortality are increased, and therapy is likely essential. Corticosteroids are commonly used at some stage, but tapering is difficult; immunosuppressive agents such as cyclosporine, mycophenolate, cyclophosphamide, and methotrexate have improved the pulmonary disease in some. Tumor necrosis factor inhibitors have also been used with benefit in some cases. Combination immunosuppression with rituximab and azathioprine or other T-cell immune-suppressing agents has been proposed and appears now to be the most commonly used combination.46 For guidance in the application of these therapies, however, hematology input is often required.
Treatment of cancer in CVID
In general, the treatment of cancer in CVID is the same as applied to immunocompetent subjects with, of course, the inclusion of standard immunoglobulin therapy.
Stem cell transplantation in CVID
There is still limited information on hematopoietic stem cell and bone marrow transplantation in CVID. A question about this procedure is likely to arise when T-cell immunity is also greatly impaired and/or life-threating complications such as lymphoma or severe infections are present. Collecting retrospective data on 25 patients with CVID (aged 8 to 50 years) who underwent transplantation for lymphoma, severe autoimmunity, refractory cytopenias, or infections, the overall survival rate was 48%; for those with lymphoma, survival was 83%. The major causes of death were graft-versus-host disease and infections. Immunoglobulin substitution was stopped in 50% of 12 surviving patients, and in 11 of these, the condition for which hematopoietic stem cell transplantation was performed was resolved.47 When hypomorphic defects of genes are identified, such as in adenosine deaminase, Artemis, RAG1 or RAG2 (recombination-activating genes), or immune dysregulation genes now found in patients labeled with CVID as discussed below, this option seems clearer. In addition, with better and/or earlier selection of subjects, results may improve.
Hematologic monitoring
Most patients with CVID carry out normal activities; many have been treated with immunoglobulin in home care programs for decades. Although these improvements represent ongoing advances in medical care, regularly scheduled and careful follow-up is mandatory, because new problems may arise or evolve over time. Stable patients can be seen at least at yearly intervals, and those with various complications can be seen at shorter intervals such as 3 to 6 months. Because many of these patients have enlarged spleens, this alone is generally not medically important, unless hypersplenism leads to cytopenias; rituximab administered at intervals of 1 to 3 years can be used to control this in some patients, but current guidelines are lacking. The issue of enlarged lymph nodes is always troublesome. When new nodes appear and persist, biopsy may be required; however, in most cases, lymphomas are extranodal and appear in unusual locations such as lung or mucosa-associated tissues and are not amenable to any standardized follow-up measures.
Acknowledgments
This work was partially supported by the National Institutes of Health under National Institute of Allergy and Infectious Diseases grants AI-061093 and AI-08603.
Authorship
Contribution: C.C.R. wrote and researched this text.
Conflict-of-interest disclosure: C.C.-R. declares no competing financial interests. Off-label drug use: None disclosed.
Correspondence: Charlotte Cunningham-Rundles, Departments of Medicine and Pediatrics, Precision Immunology Institute, Icahn School of Medicine at Mount Sinai, 1425 Madison Ave, Room 1120, New York, NY 10029; e-mail: charlotte.cunningham-rundles@mssm.edu.
This article was selected by the Blood and Hematology 2019 American Society of Hematology Education Program editors for concurrent submission to Blood and Hematology 2019. It is reprinted in Hematology Am Soc Hematol Educ Program. 2019;2019:449-456.